Anda mungkin juga menyukai
- The Essence of Success - Earl NightingaleDokumen2 halamanThe Essence of Success - Earl NightingaleDegrace Ns40% (15)
- Payroll Canadian 1st Edition Dryden Test BankDokumen38 halamanPayroll Canadian 1st Edition Dryden Test Bankriaozgas3023100% (14)
- Virgilio S. Delima V. Susan Mercaida Gois GR NO. 178352 - June 17, 2008 FactsDokumen6 halamanVirgilio S. Delima V. Susan Mercaida Gois GR NO. 178352 - June 17, 2008 FactsNikki BarenaBelum ada peringkat
- Celia Salazar-Biosystem Analysis of The Hypoxia Inducible DomainDokumen20 halamanCelia Salazar-Biosystem Analysis of The Hypoxia Inducible DomainPablo valderramaBelum ada peringkat
- Moléculas PaperDokumen44 halamanMoléculas PaperCarlla Araújo Guarani KaiowáBelum ada peringkat
- 8-OHdG DeterminationDokumen7 halaman8-OHdG Determinationapi-19762689Belum ada peringkat
- Antiageing 2Dokumen14 halamanAntiageing 2Wwwanand111Belum ada peringkat
- ChemMedChem (2009), 4 (8), 1269-1272Dokumen4 halamanChemMedChem (2009), 4 (8), 1269-1272James TianBelum ada peringkat
- Aptamers As Future DrugsDokumen25 halamanAptamers As Future Drugspsc anandBelum ada peringkat
- OncometabolitosDokumen7 halamanOncometabolitosAinia KorrsBelum ada peringkat
- Ni Hms 165359Dokumen19 halamanNi Hms 165359regiadestyBelum ada peringkat
- Helvetica Chimica Acta - 2005 - Finn - Novel Sulfonamide Derivatives As Inhibitors of Histone DeacetylaseDokumen28 halamanHelvetica Chimica Acta - 2005 - Finn - Novel Sulfonamide Derivatives As Inhibitors of Histone DeacetylaseWalid ElgammalBelum ada peringkat
- Dabelsteen1996 PDFDokumen12 halamanDabelsteen1996 PDFjahdsdjad asffdhsajhajdkBelum ada peringkat
- Neuropharmacology: Vsevolod Katritch, Irina Kufareva, Ruben AbagyanDokumen8 halamanNeuropharmacology: Vsevolod Katritch, Irina Kufareva, Ruben AbagyanSociedad Peruana De Fisiología FisiopatologíaBelum ada peringkat
- An artificial metalloenzyme for catalytic cancerDokumen7 halamanAn artificial metalloenzyme for catalytic cancerShyamal KarmakarBelum ada peringkat
- Oxidative Phosporylation in Cancer CellsDokumen9 halamanOxidative Phosporylation in Cancer CellsM Naufal IlmiBelum ada peringkat
- Cinases and DiseasesDokumen15 halamanCinases and Diseasesseb2008Belum ada peringkat
- Acs Biochem 7b00795Dokumen5 halamanAcs Biochem 7b00795Romana MasnikosaBelum ada peringkat
- Platinum Complexes As Anticancer AgentsDokumen22 halamanPlatinum Complexes As Anticancer AgentsDrs_HarsanaBelum ada peringkat
- Advances in The Design of Organometallic Anticancer ComplexesDokumen26 halamanAdvances in The Design of Organometallic Anticancer ComplexesriyonaBelum ada peringkat
- Nature 11896Dokumen10 halamanNature 11896Sameera HameedBelum ada peringkat
- 2016 HarmalolDokumen11 halaman2016 Harmaloltaoufik akabliBelum ada peringkat
- 9 Sudemycins 2011Dokumen8 halaman9 Sudemycins 2011milenerato2240Belum ada peringkat
- Small Molecule MDM2 X Inhibitors and PROTAC Degraders F - 2020 - Acta PharmaceutDokumen26 halamanSmall Molecule MDM2 X Inhibitors and PROTAC Degraders F - 2020 - Acta PharmaceutMohammed Shuaib AhmedBelum ada peringkat
- FeinbergDokumen12 halamanFeinbergBunny SmithBelum ada peringkat
- Regulation of Chromatin by Histone Modifications: ReviewDokumen15 halamanRegulation of Chromatin by Histone Modifications: ReviewFlorencia FirenzeBelum ada peringkat
- Med 21275Dokumen38 halamanMed 21275suryasanBelum ada peringkat
- Journal of Medicinal Chemistry ArticleDokumen63 halamanJournal of Medicinal Chemistry ArticleJudy UgwuegbuBelum ada peringkat
- Chi Shing Sum Et Al - Effects of N-Ethylmaleimide On Conformational Equilibria in Purified Cardiac Muscarinic ReceptorsDokumen16 halamanChi Shing Sum Et Al - Effects of N-Ethylmaleimide On Conformational Equilibria in Purified Cardiac Muscarinic ReceptorsCortate15gBelum ada peringkat
- Nat Chem - A Ring-Distortion Strategy To Const PDFDokumen8 halamanNat Chem - A Ring-Distortion Strategy To Const PDFPham Ha Thanh TungBelum ada peringkat
- Benachour and Seralini-Glyphosate Formulations Inducew Apoptosis and Necrosis in Human Umbilical, Embryonic, and Placental Cells (2009)Dokumen11 halamanBenachour and Seralini-Glyphosate Formulations Inducew Apoptosis and Necrosis in Human Umbilical, Embryonic, and Placental Cells (2009)Héctor MendozaBelum ada peringkat
- Ab_Oxidation and Deamidation of Monoclonal Antibody Products- Potential Impact on Stability Biological Activity and EfficacyDokumen16 halamanAb_Oxidation and Deamidation of Monoclonal Antibody Products- Potential Impact on Stability Biological Activity and EfficacyCher IshBelum ada peringkat
- 2020 Epigenetic Evaluation of Valproic Acid DerivativesDokumen9 halaman2020 Epigenetic Evaluation of Valproic Acid DerivativesMarco MerazBelum ada peringkat
- Review Class Iia 2019Dokumen47 halamanReview Class Iia 2019Nacido para BendcirBelum ada peringkat
- Nuclear Receptors, Chemistry Of: Advanced ArticleDokumen10 halamanNuclear Receptors, Chemistry Of: Advanced ArticleazzaassBelum ada peringkat
- Feedback Activation of LIFR Limits Response To HDAC Inh in Breast CancerDokumen16 halamanFeedback Activation of LIFR Limits Response To HDAC Inh in Breast CancerKudelko MatBelum ada peringkat
- Zac 4651Dokumen6 halamanZac 4651bondester4Belum ada peringkat
- Histone Deacetylases Transcriptional Control, and Cancer.Dokumen16 halamanHistone Deacetylases Transcriptional Control, and Cancer.jose_gladiusBelum ada peringkat
- CLASSIFICATIONAdvances and Challenges of HDACDokumen29 halamanCLASSIFICATIONAdvances and Challenges of HDACRahul JanaBelum ada peringkat
- tmp37AD TMPDokumen11 halamantmp37AD TMPFrontiersBelum ada peringkat
- E2017005 PDFDokumen6 halamanE2017005 PDFComputational and Structural Biotechnology JournalBelum ada peringkat
- 1 s2.0 S1773224721007024 MainDokumen34 halaman1 s2.0 S1773224721007024 MainNihal ChauhanBelum ada peringkat
- Colorectal Cancer Cell Lines Show Striking Diversity of Their O-Glycome Reflecting The Cellular Differentiation PhenotypeDokumen14 halamanColorectal Cancer Cell Lines Show Striking Diversity of Their O-Glycome Reflecting The Cellular Differentiation PhenotypeRapazito RagazzoBelum ada peringkat
- When Starvation Therapy Meets Chemodynamic Therapy - 2022 - ChemPhysMaterDokumen17 halamanWhen Starvation Therapy Meets Chemodynamic Therapy - 2022 - ChemPhysMaterKetua PpkBelum ada peringkat
- Aysegül Zeyno Epigenetic and Nutritient Lecture - New SunumDokumen26 halamanAysegül Zeyno Epigenetic and Nutritient Lecture - New SunumzeynoleeeBelum ada peringkat
- Discovery and Reduction of Tryptophan Oxidation-Induced IgG1 Fragmentation in A Polysorbate 80-Dependent MannerDokumen9 halamanDiscovery and Reduction of Tryptophan Oxidation-Induced IgG1 Fragmentation in A Polysorbate 80-Dependent MannerAdamBelum ada peringkat
- JMC2013 Cyclin Dependent Kinase InhibitorDokumen15 halamanJMC2013 Cyclin Dependent Kinase InhibitorVincent GeruszBelum ada peringkat
- Analysis of Rab GTPases in Dictyostelium discoideumDokumen34 halamanAnalysis of Rab GTPases in Dictyostelium discoideumdavid_stephens_29Belum ada peringkat
- The Metabolic Role of AMPK in Cancer Multi-Drug ResistanceDokumen60 halamanThe Metabolic Role of AMPK in Cancer Multi-Drug ResistanceAnişoara FrunzeBelum ada peringkat
- 111480Dokumen7 halaman111480VYAPAR INDIABelum ada peringkat
- Synthesis and Antifilarial Evaluation of N, N - Xylofuranosylated DiaminoalkanesDokumen12 halamanSynthesis and Antifilarial Evaluation of N, N - Xylofuranosylated DiaminoalkanesRumana AhmadBelum ada peringkat
- Article 1Dokumen15 halamanArticle 1Dickson PaschalSospeterBelum ada peringkat
- Research ArticleDokumen9 halamanResearch ArticleMinh Đức HoàngBelum ada peringkat
- Factor Viia Inhibitors: Improved Pharmacokinetic Parameters: Table 1Dokumen4 halamanFactor Viia Inhibitors: Improved Pharmacokinetic Parameters: Table 1ebi1364Belum ada peringkat
- Giacci A 2016Dokumen9 halamanGiacci A 2016yagami19lightBelum ada peringkat
- Protein DegradationDokumen16 halamanProtein DegradationBhakti HiraniBelum ada peringkat
- Article 1 PDFDokumen19 halamanArticle 1 PDFAnonymous 5bgTK7qzsBelum ada peringkat
- Analytical Biochemistry: Swathi Krishnan, Evys Collazo, Patricia A. Ortiz-Tello, Raymond C. TrievelDokumen6 halamanAnalytical Biochemistry: Swathi Krishnan, Evys Collazo, Patricia A. Ortiz-Tello, Raymond C. TrievelTidar MelianiBelum ada peringkat
- 2010 - Unfolding Free Energy of A Two-Domain - Transmembrane Sugar Transport ProteinDokumen6 halaman2010 - Unfolding Free Energy of A Two-Domain - Transmembrane Sugar Transport Protein华贝杰Belum ada peringkat
- Coordination Chemistry—XVI: XVIth International Conference on Coordination ChemistryDari EverandCoordination Chemistry—XVI: XVIth International Conference on Coordination ChemistryBelum ada peringkat
- Analytical Characterization of BiotherapeuticsDari EverandAnalytical Characterization of BiotherapeuticsJennie R. LillBelum ada peringkat
- Metabolite Safety in Drug DevelopmentDari EverandMetabolite Safety in Drug DevelopmentSuzanne L. IversonBelum ada peringkat
- Review04 - Natural Product Reports (2012), 29, 449-456Dokumen8 halamanReview04 - Natural Product Reports (2012), 29, 449-456James TianBelum ada peringkat
- Review03-Angewandte Chemie, International Edition (2010), 49 (50), 9592-9628Dokumen37 halamanReview03-Angewandte Chemie, International Edition (2010), 49 (50), 9592-9628James TianBelum ada peringkat
- Blood, (2012), 119 (4), 1008-1017Dokumen11 halamanBlood, (2012), 119 (4), 1008-1017James TianBelum ada peringkat
- Molecules (2011), 16 4681-4694Dokumen14 halamanMolecules (2011), 16 4681-4694James TianBelum ada peringkat
- Review01-Natural Product Reports (2010), 27 (8), 1186-1203Dokumen18 halamanReview01-Natural Product Reports (2010), 27 (8), 1186-1203James TianBelum ada peringkat
- Journal of Organic Chemistry (2011), 76 (4), 1140-1150Dokumen11 halamanJournal of Organic Chemistry (2011), 76 (4), 1140-1150James TianBelum ada peringkat
- Largazole AnaloguesDokumen26 halamanLargazole AnaloguesJames TianBelum ada peringkat
- Review02-Natural Product Reports (2009), 26 (10), 1293-1320Dokumen28 halamanReview02-Natural Product Reports (2009), 26 (10), 1293-1320James TianBelum ada peringkat
- Largazole HDAC SARDokumen25 halamanLargazole HDAC SARJames TianBelum ada peringkat
- PLoS One, (2012), 7 (1), 1-9Dokumen9 halamanPLoS One, (2012), 7 (1), 1-9James TianBelum ada peringkat
- Journal of The American Chemical Society (2011), 133 (32), 12474-12477Dokumen4 halamanJournal of The American Chemical Society (2011), 133 (32), 12474-12477James TianBelum ada peringkat
- Journal of Asian Natural Products Research, (2010), 12 (11), 940-949Dokumen11 halamanJournal of Asian Natural Products Research, (2010), 12 (11), 940-949James TianBelum ada peringkat
- Journal of Pharmacology and Experimental Therapeutics (2010), 335 (2), 351-361-SuppDokumen12 halamanJournal of Pharmacology and Experimental Therapeutics (2010), 335 (2), 351-361-SuppJames TianBelum ada peringkat
- ACS Medicinal Chemistry Letters (2011), 2 (3), 248-251Dokumen4 halamanACS Medicinal Chemistry Letters (2011), 2 (3), 248-251James TianBelum ada peringkat
- Organic Letters (2010), 12 (6), 1368-1371Dokumen4 halamanOrganic Letters (2010), 12 (6), 1368-1371James TianBelum ada peringkat
- Journal of Medicinal Chemistry (2010), 53 (12), 4654-4667Dokumen14 halamanJournal of Medicinal Chemistry (2010), 53 (12), 4654-4667James TianBelum ada peringkat
- Synthesis (2009), (17), 2873-2880Dokumen8 halamanSynthesis (2009), (17), 2873-2880James TianBelum ada peringkat
- Journal of Pharmacology and Experimental Therapeutics (2010), 335 (2), 351-361Dokumen11 halamanJournal of Pharmacology and Experimental Therapeutics (2010), 335 (2), 351-361James TianBelum ada peringkat
- ChemBioChem (2009), 10 (10), 1634-1639Dokumen6 halamanChemBioChem (2009), 10 (10), 1634-1639James TianBelum ada peringkat
- Synlett (2008), (15), 2379-2383Dokumen5 halamanSynlett (2008), (15), 2379-2383James TianBelum ada peringkat
- Journal of Medicinal Chemistry (2009), 52 (8), 2163-2176Dokumen14 halamanJournal of Medicinal Chemistry (2009), 52 (8), 2163-2176James TianBelum ada peringkat
- Organic Letters (2008), 10 (18), 4021-4024Dokumen4 halamanOrganic Letters (2008), 10 (18), 4021-4024James TianBelum ada peringkat
- Organic Letters (2009), 11 (6), 1301-1304Dokumen4 halamanOrganic Letters (2009), 11 (6), 1301-1304James TianBelum ada peringkat
- Angewandte Chemie, International Edition (2008), 47 (34), 6483-6485Dokumen3 halamanAngewandte Chemie, International Edition (2008), 47 (34), 6483-6485James TianBelum ada peringkat
- Organic Letters (2008), 10 (16), 3595-3598Dokumen4 halamanOrganic Letters (2008), 10 (16), 3595-3598James TianBelum ada peringkat
- Journal of The American Chemical Society (2009), 131 (8), 2900-2905Dokumen6 halamanJournal of The American Chemical Society (2009), 131 (8), 2900-2905James TianBelum ada peringkat
- Organic Letters (2008), 10 (17), 3907-3909Dokumen3 halamanOrganic Letters (2008), 10 (17), 3907-3909James TianBelum ada peringkat
- Journal of The American Chemical Society (2008), 130 (33), 11219-11222Dokumen4 halamanJournal of The American Chemical Society (2008), 130 (33), 11219-11222James TianBelum ada peringkat
- Oscar Ortega Lopez - 1.2.3.a BinaryNumbersConversionDokumen6 halamanOscar Ortega Lopez - 1.2.3.a BinaryNumbersConversionOscar Ortega LopezBelum ada peringkat
- Master StationDokumen138 halamanMaster StationWilmer Quishpe AndradeBelum ada peringkat
- Cantilever Retaining Wall AnalysisDokumen7 halamanCantilever Retaining Wall AnalysisChub BokingoBelum ada peringkat
- KL Wellness City LIvewell 360 2023Dokumen32 halamanKL Wellness City LIvewell 360 2023tan sietingBelum ada peringkat
- 04 Activity 2Dokumen2 halaman04 Activity 2Jhon arvie MalipolBelum ada peringkat
- The Non Technical Part: Sample Interview Questions For Network EngineersDokumen5 halamanThe Non Technical Part: Sample Interview Questions For Network EngineersblablaBelum ada peringkat
- Final Project Report: Uop, LLCDokumen165 halamanFinal Project Report: Uop, LLCSiddharth KishanBelum ada peringkat
- Cagayan Electric Company v. CIRDokumen2 halamanCagayan Electric Company v. CIRCocoyPangilinanBelum ada peringkat
- Affordable Care Act Tax - Fact CheckDokumen26 halamanAffordable Care Act Tax - Fact CheckNag HammadiBelum ada peringkat
- Duct Design ChartDokumen7 halamanDuct Design ChartMohsen HassanBelum ada peringkat
- Sierra Wireless AirPrimeDokumen2 halamanSierra Wireless AirPrimeAminullah -Belum ada peringkat
- De Thi Thu THPT Quoc Gia Mon Tieng Anh Truong THPT Hai An Hai Phong Nam 2015Dokumen10 halamanDe Thi Thu THPT Quoc Gia Mon Tieng Anh Truong THPT Hai An Hai Phong Nam 2015nguyen ngaBelum ada peringkat
- SEEPZ Special Economic ZoneDokumen2 halamanSEEPZ Special Economic ZonetarachandmaraBelum ada peringkat
- Iqvia PDFDokumen1 halamanIqvia PDFSaksham DabasBelum ada peringkat
- BILL OF SALE Pre ApproveDokumen1 halamanBILL OF SALE Pre ApprovedidinurieliaBelum ada peringkat
- Organization Structure GuideDokumen6 halamanOrganization Structure GuideJobeth BedayoBelum ada peringkat
- Incident Report Form: RPSG-IMS-F-24 Accident and Investigation Form 5ADokumen2 halamanIncident Report Form: RPSG-IMS-F-24 Accident and Investigation Form 5ARocky BisBelum ada peringkat
- Lab - Activity CCNA 2 Exp: 7.5.3Dokumen13 halamanLab - Activity CCNA 2 Exp: 7.5.3Rico Agung FirmansyahBelum ada peringkat
- ETP Research Proposal Group7 NewDokumen12 halamanETP Research Proposal Group7 NewlohBelum ada peringkat
- Ridge Regression: A Concise GuideDokumen132 halamanRidge Regression: A Concise GuideprinceBelum ada peringkat
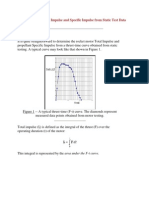
- Determining Total Impulse and Specific Impulse From Static Test DataDokumen4 halamanDetermining Total Impulse and Specific Impulse From Static Test Datajai_selvaBelum ada peringkat
- Research Grants Final/Terminal/Exit Progress Report: Instructions and Reporting FormDokumen13 halamanResearch Grants Final/Terminal/Exit Progress Report: Instructions and Reporting FormBikaZee100% (1)
- Liability WaiverDokumen1 halamanLiability WaiverTop Flight FitnessBelum ada peringkat
- Parasim CADENCEDokumen166 halamanParasim CADENCEvpsampathBelum ada peringkat
- Nammo Bulletin 2014Dokumen13 halamanNammo Bulletin 2014Dmitry Karpov0% (1)
- University Assignment Report CT7098Dokumen16 halamanUniversity Assignment Report CT7098Shakeel ShahidBelum ada peringkat
- VB 2Dokumen11 halamanVB 2Sudhir IkkeBelum ada peringkat