Anda mungkin juga menyukai
- Complementary and Alternative Medical Lab Testing Part 2: RespiratoryDari EverandComplementary and Alternative Medical Lab Testing Part 2: RespiratoryBelum ada peringkat
- The Spectrum Evan SydromDokumen4 halamanThe Spectrum Evan SydromMohankummar MuniandyBelum ada peringkat
- Evans Syndrome - StatPearls - NCBI BookshelfDokumen4 halamanEvans Syndrome - StatPearls - NCBI BookshelfJunbek nov23Belum ada peringkat
- 456 FullDokumen4 halaman456 FullDino GoncalvesBelum ada peringkat
- Laboratory Features of Systemic Lupus Erythematosus (SLE)Dokumen6 halamanLaboratory Features of Systemic Lupus Erythematosus (SLE)AmrKamalBelum ada peringkat
- Complications of Infectious Mononucleosis in Children: PediatricsDokumen10 halamanComplications of Infectious Mononucleosis in Children: PediatricsOrhan ErBelum ada peringkat
- HiperesosinofiliaDokumen3 halamanHiperesosinofiliaAlifa Hasya NadhiraBelum ada peringkat
- 1546 0096 5 7 PDFDokumen4 halaman1546 0096 5 7 PDFArdanta Dat Topik TariganBelum ada peringkat
- FIX Jurnal Reading SJS-TENDokumen14 halamanFIX Jurnal Reading SJS-TENFarah ArdindaBelum ada peringkat
- Ana 29 492Dokumen6 halamanAna 29 492AdilZyaniBelum ada peringkat
- Epidemiology of Paediatric Immune Thrombocytopenia in The General Practice Research DatabaseDokumen1 halamanEpidemiology of Paediatric Immune Thrombocytopenia in The General Practice Research DatabaseSuardimanAchoBelum ada peringkat
- Original Article: Pulmonary Tuberculosis in Childhood Nephrotic Syndrome (A Cross Sectional Study)Dokumen5 halamanOriginal Article: Pulmonary Tuberculosis in Childhood Nephrotic Syndrome (A Cross Sectional Study)Maghfirah RahimaBelum ada peringkat
- Special Report 2010.xDokumen15 halamanSpecial Report 2010.xDianMuliasariBelum ada peringkat
- Antiphospholipid Syndrome (Aps) and PregnancyDokumen36 halamanAntiphospholipid Syndrome (Aps) and Pregnancyskeisham11Belum ada peringkat
- Parvovirus Lupus PDFDokumen3 halamanParvovirus Lupus PDFDaniel PintoBelum ada peringkat
- Systemic Lupus Erythematosus: Authors: DR Jessica J Manson and DR Anisur RahmanDokumen0 halamanSystemic Lupus Erythematosus: Authors: DR Jessica J Manson and DR Anisur RahmanRizka Norma WiwekaBelum ada peringkat
- Differences in Disease Phenotype and Severity in SLE Across Age GroupsDokumen9 halamanDifferences in Disease Phenotype and Severity in SLE Across Age GroupsenilBelum ada peringkat
- Henoch Schonlein Purpura (IgA Vasculitis)Dokumen15 halamanHenoch Schonlein Purpura (IgA Vasculitis)Emily Eresuma100% (1)
- Acta PediatricaDokumen6 halamanActa PediatricafelipeortizrBelum ada peringkat
- Respiratory Syncytial Virus Infections in Children With Acute Myeloid Leukemia: A Report From The Children's Oncology GroupDokumen3 halamanRespiratory Syncytial Virus Infections in Children With Acute Myeloid Leukemia: A Report From The Children's Oncology GrouperickmattosBelum ada peringkat
- Electrolyte Imbalances in An Unselected Population in An Emergency Department: A Retrospective Cohort StudyDokumen14 halamanElectrolyte Imbalances in An Unselected Population in An Emergency Department: A Retrospective Cohort StudyVincent ReyesBelum ada peringkat
- Kejang HiperglikemiaDokumen10 halamanKejang HiperglikemiaAbigail PheiliaBelum ada peringkat
- Research Article: Comparative Assessment of Cytokine Pattern in Early and Late Onset of Neonatal SepsisDokumen9 halamanResearch Article: Comparative Assessment of Cytokine Pattern in Early and Late Onset of Neonatal SepsisPutri HolmesBelum ada peringkat
- Jurnal HepatitisDokumen5 halamanJurnal HepatitislevyBelum ada peringkat
- Pediatric Systemic Lupus Erythematosus Associated With AutoimmuneDokumen9 halamanPediatric Systemic Lupus Erythematosus Associated With AutoimmunegistaluvikaBelum ada peringkat
- Systemic Lupus ErythematosusDokumen10 halamanSystemic Lupus ErythematosuszkxxyyBelum ada peringkat
- SLE DR - Nita Uul PunyaDokumen18 halamanSLE DR - Nita Uul PunyaduratulkhBelum ada peringkat
- Case Report: Acute Hepatitis E in A Pediatric Traveler Presenting With Features of Autoimmune Hepatitis: A Diagnostic and Therapeutic ChallengeDokumen4 halamanCase Report: Acute Hepatitis E in A Pediatric Traveler Presenting With Features of Autoimmune Hepatitis: A Diagnostic and Therapeutic Challengeanjingbasah 24Belum ada peringkat
- Eritema Nodoso: Causas Más Prevalentes en Pacientes Que Se Hospitalizan para Estudio, y Recomendaciones para El DiagnósticoDokumen7 halamanEritema Nodoso: Causas Más Prevalentes en Pacientes Que Se Hospitalizan para Estudio, y Recomendaciones para El DiagnósticoCamilo PerillaBelum ada peringkat
- Referat Status OkDokumen34 halamanReferat Status OkmarfirarizkiBelum ada peringkat
- Nazer Prognostic Index For Wilsons DiseaseDokumen5 halamanNazer Prognostic Index For Wilsons Diseasepurushotham ABelum ada peringkat
- Juvenile Lupus Erythematosus: Fourteen Years of Experience: Juvenil Lupus Eritematozus: On Dört Yıllık DeneyimDokumen8 halamanJuvenile Lupus Erythematosus: Fourteen Years of Experience: Juvenil Lupus Eritematozus: On Dört Yıllık DeneyimRahman SetiawanBelum ada peringkat
- Epidemiological Study of Clinical and Laboratory Profiles of Patients Woth ALL at DR SoetomoDokumen5 halamanEpidemiological Study of Clinical and Laboratory Profiles of Patients Woth ALL at DR SoetomoJulia Intan Permata SariBelum ada peringkat
- Kejang Hiperglikemi PDFDokumen10 halamanKejang Hiperglikemi PDFNishfullaili Nurun NisaBelum ada peringkat
- Plaquetas 2Dokumen28 halamanPlaquetas 2Gustavo CasanovaBelum ada peringkat
- Practice: Kikuchi-Fujimoto Disease: Lymphadenopathy in SiblingsDokumen4 halamanPractice: Kikuchi-Fujimoto Disease: Lymphadenopathy in SiblingsBobby Faisyal RakhmanBelum ada peringkat
- Guidelines On The Investigation and Management of Antiphospholipid SyndromeDokumen12 halamanGuidelines On The Investigation and Management of Antiphospholipid Syndromefarish_machyaBelum ada peringkat
- Retrospective Analysis of Deaths Due To Drug - Induced StevensJohnson Syndrome (SJS) and Toxic Epidermal Necrolysis (TEN) in Inpatients Admitted in The Dermatology Unit of A Tertiary Care HospitalDokumen4 halamanRetrospective Analysis of Deaths Due To Drug - Induced StevensJohnson Syndrome (SJS) and Toxic Epidermal Necrolysis (TEN) in Inpatients Admitted in The Dermatology Unit of A Tertiary Care HospitalIOSRjournalBelum ada peringkat
- Extraintestinal Focal Infections in Adults With Nontyphoid Salmonella 2006Dokumen10 halamanExtraintestinal Focal Infections in Adults With Nontyphoid Salmonella 2006Carlos Antonio Arrieta NieblesBelum ada peringkat
- jcm00141 0073Dokumen6 halamanjcm00141 0073Ibrahim QariBelum ada peringkat
- Ann Rheum Dis 2002 Rudwaleit 968 74Dokumen8 halamanAnn Rheum Dis 2002 Rudwaleit 968 74Kobayashi MaruBelum ada peringkat
- Acr2 2 74 2Dokumen5 halamanAcr2 2 74 2F-For-Fun sBelum ada peringkat
- Ovadia 2007Dokumen7 halamanOvadia 2007uditBelum ada peringkat
- High DoseDokumen6 halamanHigh DoseClaudia KosztelnikBelum ada peringkat
- Complications of Bacteriologically Confirmed Typhoid Fever in ChildrenDokumen7 halamanComplications of Bacteriologically Confirmed Typhoid Fever in Childrenfauzia azzahraBelum ada peringkat
- Current Strategies For Phenotyping and Managing Asthma in Preschool Children 2022Dokumen8 halamanCurrent Strategies For Phenotyping and Managing Asthma in Preschool Children 2022Veronica Ofelia Morachimo GarcíaBelum ada peringkat
- 1476 1503 1 PB PDFDokumen8 halaman1476 1503 1 PB PDFArief GusmanBelum ada peringkat
- A Cohort Study To Assess The New WHO Japanese Encephalitis Surveillance StandardsDokumen9 halamanA Cohort Study To Assess The New WHO Japanese Encephalitis Surveillance StandardsarmankoassracunBelum ada peringkat
- Age As A Risk Factor of Relapse Occurrence in Acute Lymphoblastic Leukemia-L1 (All-L1) in ChildrenDokumen4 halamanAge As A Risk Factor of Relapse Occurrence in Acute Lymphoblastic Leukemia-L1 (All-L1) in ChildrenYuliana WiralestariBelum ada peringkat
- American J Hematol - 2019 - Hansen - Evans Syndrome in Adults Incidence Prevalence and Survival in A Nationwide CohortDokumen11 halamanAmerican J Hematol - 2019 - Hansen - Evans Syndrome in Adults Incidence Prevalence and Survival in A Nationwide CohortFitri AudiniahBelum ada peringkat
- Lazy Leucocyte SyndromeDokumen2 halamanLazy Leucocyte SyndromeDragos BourosBelum ada peringkat
- Reye Syndrome: Clinical Manifestations and Laboratory FindingsDokumen3 halamanReye Syndrome: Clinical Manifestations and Laboratory FindingsMario HBBelum ada peringkat
- Seronegative Antiphospholipid SyndromeDokumen3 halamanSeronegative Antiphospholipid SyndromealexandruBelum ada peringkat
- Pi Is 0168827803002356Dokumen2 halamanPi Is 0168827803002356JeanetteBelum ada peringkat
- Acute Liver Failure in Cuban ChildrenDokumen7 halamanAcute Liver Failure in Cuban Childrennevermore11Belum ada peringkat
- Juvenile Rheumatoid ArthritisDokumen12 halamanJuvenile Rheumatoid ArthritisCatherine PoBelum ada peringkat
- AIHA ArticleDokumen7 halamanAIHA ArticlegffghBelum ada peringkat
- Prognostic Variables in Newly Diagnosed Childhood Immune ThrombocytopeniaDokumen5 halamanPrognostic Variables in Newly Diagnosed Childhood Immune Thrombocytopeniadhania patraBelum ada peringkat
- Henoch-Schonlein PurpuraDokumen5 halamanHenoch-Schonlein PurpuraPramita Pramana100% (1)
- Penatalaksanaan StrokeDokumen15 halamanPenatalaksanaan StrokerizeviBelum ada peringkat
- Neonatal Herpes Simplex Virus InfectionDokumen2 halamanNeonatal Herpes Simplex Virus InfectionrizeviBelum ada peringkat
- Abortion PDFDokumen3 halamanAbortion PDFrizeviBelum ada peringkat
- Meningitis PDFDokumen8 halamanMeningitis PDFrizeviBelum ada peringkat
- Evans SyndromeDokumen13 halamanEvans SyndromerizeviBelum ada peringkat
- Seizures & Stroke Risk: Brain Injury and General Rehabilitation Information SeriesDokumen8 halamanSeizures & Stroke Risk: Brain Injury and General Rehabilitation Information SeriesIli DiyanaBelum ada peringkat
- Bab Iv Daftar Pustaka: Anak, Unit Kerja Pulmonologi PP IDAI, Jakarta, Halaman 54-56Dokumen1 halamanBab Iv Daftar Pustaka: Anak, Unit Kerja Pulmonologi PP IDAI, Jakarta, Halaman 54-56rizeviBelum ada peringkat
- Seizures & Stroke Risk: Brain Injury and General Rehabilitation Information SeriesDokumen8 halamanSeizures & Stroke Risk: Brain Injury and General Rehabilitation Information SeriesIli DiyanaBelum ada peringkat
- Law MCQ 25Dokumen3 halamanLaw MCQ 25nonoBelum ada peringkat
- Phrasal Verbs en Inglés.Dokumen2 halamanPhrasal Verbs en Inglés.David Alexander Palomo QuirozBelum ada peringkat
- Taewoo Kim Et Al. v. Jump TradingDokumen44 halamanTaewoo Kim Et Al. v. Jump TradingCrainsChicagoBusiness100% (1)
- MY-SDK-10000-EE-005 - Method Statement For Concrete Pole Installation - GVB Rev1Dokumen7 halamanMY-SDK-10000-EE-005 - Method Statement For Concrete Pole Installation - GVB Rev1Seeths NairBelum ada peringkat
- Reflection Frog 1Dokumen3 halamanReflection Frog 1mariamBelum ada peringkat
- EverServ 7700 M77XX Quick Reference GuideDokumen2 halamanEverServ 7700 M77XX Quick Reference GuidetangocharliepdxBelum ada peringkat
- Practice Problems For Modulus and Logarithm Section-I: FiitjeeDokumen8 halamanPractice Problems For Modulus and Logarithm Section-I: FiitjeePratham SharmaBelum ada peringkat
- Grammar and Oral Language Development (GOLD) : Reported By: Melyn A. Bacolcol Kate Batac Julie Ann OcampoDokumen17 halamanGrammar and Oral Language Development (GOLD) : Reported By: Melyn A. Bacolcol Kate Batac Julie Ann Ocampoclara dupitasBelum ada peringkat
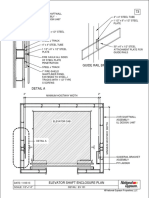
- Guide Rail Bracket AssemblyDokumen1 halamanGuide Rail Bracket AssemblyPrasanth VarrierBelum ada peringkat
- Glossario - GETTY - IngDokumen24 halamanGlossario - GETTY - IngFabio ZarattiniBelum ada peringkat
- Repro Indo China Conf PDFDokumen16 halamanRepro Indo China Conf PDFPavit KaurBelum ada peringkat
- Esp 1904 A - 70 TPH o & M ManualDokumen50 halamanEsp 1904 A - 70 TPH o & M Manualpulakjaiswal85Belum ada peringkat
- Bangalore Escorts Services - Riya ShettyDokumen11 halamanBangalore Escorts Services - Riya ShettyRiya ShettyBelum ada peringkat
- Acm Queue PDFDokumen12 halamanAcm Queue PDFShubham Anil ShahareBelum ada peringkat
- Checklist Code ReviewDokumen2 halamanChecklist Code ReviewTrang Đỗ Thu100% (1)
- Electrical System in AENDokumen21 halamanElectrical System in AENNilesh PatilBelum ada peringkat
- A Modified Linear Programming Method For Distribution System ReconfigurationDokumen6 halamanA Modified Linear Programming Method For Distribution System Reconfigurationapi-3697505Belum ada peringkat
- LOMA FLMI CoursesDokumen4 halamanLOMA FLMI CoursesCeleste Joy C. LinsanganBelum ada peringkat
- SAP IAG Admin GuideDokumen182 halamanSAP IAG Admin GuidegadesigerBelum ada peringkat
- VHP Series Five P9394Gsi S5: With Esm2 and Empact Emission Control SystemDokumen2 halamanVHP Series Five P9394Gsi S5: With Esm2 and Empact Emission Control SystemGabrielito PachacamaBelum ada peringkat
- Chapter-4 Conditional and Iterative Statements in PythonDokumen30 halamanChapter-4 Conditional and Iterative Statements in Pythonashishiet100% (1)
- SITXWHS001 - Participate in Safe Work Practices Student GuideDokumen42 halamanSITXWHS001 - Participate in Safe Work Practices Student GuideMarianne FernandoBelum ada peringkat
- 2062 TSSR Site Sharing - Rev02Dokumen44 halaman2062 TSSR Site Sharing - Rev02Rio DefragBelum ada peringkat
- Course Specifications: Fire Investigation and Failure Analysis (E901313)Dokumen2 halamanCourse Specifications: Fire Investigation and Failure Analysis (E901313)danateoBelum ada peringkat
- Upend RA Kumar: Master List of Approved Vendors For Manufacture and Supply of Electrical ItemsDokumen42 halamanUpend RA Kumar: Master List of Approved Vendors For Manufacture and Supply of Electrical Itemssantosh iyerBelum ada peringkat
- Hung201 PDFDokumen14 halamanHung201 PDFMua Dong Tuyet RoiBelum ada peringkat
- Presentacion Peaks Rms Lufs Como Usar Medidores FavorDokumen16 halamanPresentacion Peaks Rms Lufs Como Usar Medidores Favorhector.obregon.martinezBelum ada peringkat
- WP05 - ACT 01 - Development 1909Dokumen53 halamanWP05 - ACT 01 - Development 1909ramesh9966Belum ada peringkat
- Grade 10 LP Thin LensDokumen6 halamanGrade 10 LP Thin LensBrena PearlBelum ada peringkat
- Applications of Remote Sensing and Gis For UrbanDokumen47 halamanApplications of Remote Sensing and Gis For UrbanKashan Ali KhanBelum ada peringkat