Anda mungkin juga menyukai
- Pharmacokinetic & PharmacodynamicDokumen77 halamanPharmacokinetic & Pharmacodynamicdimasscrib100% (1)
- Altered Pharmacokinetics in Liver DiseasesDokumen30 halamanAltered Pharmacokinetics in Liver DiseasesNailaAns100% (1)
- Questions - Answer Bassam QQ 1Dokumen14 halamanQuestions - Answer Bassam QQ 1Bassam SaifBelum ada peringkat
- PharmacokineticsDokumen8 halamanPharmacokineticsPinay YaunBelum ada peringkat
- Drugs and Doping in SportDokumen39 halamanDrugs and Doping in SportAbhijit Sharma100% (1)
- Drug Interaction Can Be Defined As TheDokumen34 halamanDrug Interaction Can Be Defined As TheIstianah Es100% (1)
- Absorption of DrugsDokumen41 halamanAbsorption of DrugsSibtain100% (2)
- Preoperative AssessmentDokumen9 halamanPreoperative Assessment1234chocoBelum ada peringkat
- Bioavailability and First Pass MetabolismDokumen37 halamanBioavailability and First Pass MetabolismFitrye Yhana Rayyi KurniawanBelum ada peringkat
- Hydrophilic-Lipophilic BalanceDokumen56 halamanHydrophilic-Lipophilic Balancealanmarr100% (1)
- Katzung Chapter 3Dokumen2 halamanKatzung Chapter 3geldevera100% (1)
- نم هدمتعم و هعجارم اهنل اهب قوثوم لخادلاب تامولعملا Certified fromDokumen20 halamanنم هدمتعم و هعجارم اهنل اهب قوثوم لخادلاب تامولعملا Certified fromMedhat EyadaBelum ada peringkat
- Final PharmacologyDokumen284 halamanFinal PharmacologytrialqwBelum ada peringkat
- Pharmacology in Drug Discovery: Understanding Drug ResponseDari EverandPharmacology in Drug Discovery: Understanding Drug ResponseBelum ada peringkat
- Biopharmaceutics - BrahmankarDokumen414 halamanBiopharmaceutics - Brahmankarmatin586% (57)
- Principles of Pharmacology Chapter 1Dokumen37 halamanPrinciples of Pharmacology Chapter 1Muhammad ZakriaBelum ada peringkat
- BiopharmaceuticsDokumen52 halamanBiopharmaceuticsDharma ShantiniBelum ada peringkat
- Dosage and Solutions - FinalDokumen27 halamanDosage and Solutions - Finalramoli1988100% (1)
- Clinical PharmacologDokumen81 halamanClinical PharmacologSHILOTABelum ada peringkat
- Drug Metabolism in DiseasesDari EverandDrug Metabolism in DiseasesWen XieBelum ada peringkat
- Handbook of Drug Interaction and the Mechanism of InteractionDari EverandHandbook of Drug Interaction and the Mechanism of InteractionPenilaian: 1 dari 5 bintang1/5 (1)
- Pharmacology: Pharmacokinetic & Dose: Ana Khusnul Faizah Farmasi FK Uht 2018Dokumen27 halamanPharmacology: Pharmacokinetic & Dose: Ana Khusnul Faizah Farmasi FK Uht 2018Aulia rahmawatiBelum ada peringkat
- Abdullahi V Pfizer PaperDokumen2 halamanAbdullahi V Pfizer PaperTeam2KissBelum ada peringkat
- List of Indus of Pithampur ExcelDokumen32 halamanList of Indus of Pithampur ExcelWHATSAPP VIRAL & FUNNY COMEDY MAST WALE VIDEOS100% (1)
- Metabolism, Pharmacokinetics and Toxicity of Functional GroupsDokumen545 halamanMetabolism, Pharmacokinetics and Toxicity of Functional GroupsDenisa Nițu100% (1)
- Language of PreventionDokumen9 halamanLanguage of Prevention1234chocoBelum ada peringkat
- Bioavailability and First Pass ClearanceDokumen3 halamanBioavailability and First Pass ClearanceMergu Bala RajuBelum ada peringkat
- First Pass MetabolismDokumen8 halamanFirst Pass MetabolismMuhammad bilalBelum ada peringkat
- PharmacologyDokumen48 halamanPharmacologyRere Oslec100% (1)
- Assignment On Pharmacokinetic Drug Interaction: Teerthanker Mahaveer UniversityDokumen8 halamanAssignment On Pharmacokinetic Drug Interaction: Teerthanker Mahaveer Universityt24008882Belum ada peringkat
- M.Pharm (PH - Ceutics) : Presented byDokumen28 halamanM.Pharm (PH - Ceutics) : Presented byDipankar DasBelum ada peringkat
- Ayine Chap 4Dokumen59 halamanAyine Chap 4Abdi JifarBelum ada peringkat
- Assignment PharmaDokumen10 halamanAssignment Pharmac2bmqsfkp7Belum ada peringkat
- Ceutics CAT 2Dokumen9 halamanCeutics CAT 2Ria KitlerBelum ada peringkat
- Bioavailability: Clinical Research TrialsDokumen2 halamanBioavailability: Clinical Research TrialsRohan SawantBelum ada peringkat
- Makalah KBM Kel 3Dokumen32 halamanMakalah KBM Kel 3Novia RozadiBelum ada peringkat
- 3 - Pharmakcokinetics & Pharmacodynamics: Apparent VolumeDokumen10 halaman3 - Pharmakcokinetics & Pharmacodynamics: Apparent VolumexCadisRaiBelum ada peringkat
- Drug InteractionsDokumen43 halamanDrug InteractionsNinaBelum ada peringkat
- Pharmaco KineticsDokumen10 halamanPharmaco KineticsVithyaah ParameswaranBelum ada peringkat
- 03 - Principles of PharmacokineticsDokumen25 halaman03 - Principles of PharmacokineticsSimonBelum ada peringkat
- Basic Pharmacological Principles - WSAVA2011 - VINDokumen13 halamanBasic Pharmacological Principles - WSAVA2011 - VINMystry MailBelum ada peringkat
- General Pharmacology NotesDokumen76 halamanGeneral Pharmacology NotesXara Barretto Magadia100% (1)
- Trepanier2013 Applying Pharmacokinetics To Veterinary Clinical PracticeDokumen14 halamanTrepanier2013 Applying Pharmacokinetics To Veterinary Clinical PracticeSarahBelum ada peringkat
- Bioavailability - A Pharmaceutical ReviewDokumen17 halamanBioavailability - A Pharmaceutical Reviewraju155940550% (2)
- Pharmacokinetic and Pharmacodynamic2021Dokumen12 halamanPharmacokinetic and Pharmacodynamic2021STEFANIA AYA DIAZBelum ada peringkat
- Absorption WindowsDokumen9 halamanAbsorption WindowsFilip IlievskiBelum ada peringkat
- Select The Single Best Answer: A. BioavailabilityDokumen14 halamanSelect The Single Best Answer: A. BioavailabilityleahbayBelum ada peringkat
- A Dictionary of Terms Used in Drug MetabolismDokumen7 halamanA Dictionary of Terms Used in Drug MetabolismOksana KamenetskaBelum ada peringkat
- Research ArticleDokumen12 halamanResearch ArticleMary VegaBelum ada peringkat
- PDQ PharmaDokumen17 halamanPDQ PharmaRui PenedaBelum ada peringkat
- Facttors Affecting Drug MetabolismDokumen3 halamanFacttors Affecting Drug MetabolismAnanyaBelum ada peringkat
- Bio Case StudyDokumen20 halamanBio Case StudyMd. Nure Alamin SiddikBelum ada peringkat
- Biopharmaceutics KU (Maam Shaista)Dokumen13 halamanBiopharmaceutics KU (Maam Shaista)areebaBelum ada peringkat
- Farmacologia y Farmacocinetica NeonatalDokumen8 halamanFarmacologia y Farmacocinetica NeonatalRafael Flores PintoBelum ada peringkat
- Drug - Drug Int2Dokumen35 halamanDrug - Drug Int2alishba100% (2)
- College of Pharmacy Biopharmaceutics and Pharmacokinetics Assignment No. 1Dokumen6 halamanCollege of Pharmacy Biopharmaceutics and Pharmacokinetics Assignment No. 1ERIKA JADE TORRESBelum ada peringkat
- Pha 201 Lecture NoteDokumen10 halamanPha 201 Lecture NoteDebby14Belum ada peringkat
- Food Effects On Drug Bioavailability: Implications For New and Generic Drug DevelopmentDokumen19 halamanFood Effects On Drug Bioavailability: Implications For New and Generic Drug DevelopmentFanny MoralesBelum ada peringkat
- Pharmacology: Core Curriculum in NephrologyDokumen11 halamanPharmacology: Core Curriculum in NephrologyYuppie RajBelum ada peringkat
- Chapter 2Dokumen13 halamanChapter 2simbaiBelum ada peringkat
- Biopharmaceutics by John G. WagnerDokumen29 halamanBiopharmaceutics by John G. WagnerNick GanteBelum ada peringkat
- Review Article Dose-Dependent Pharmacokinetics: Experimental Observations and Theoretical ConsiderationsDokumen31 halamanReview Article Dose-Dependent Pharmacokinetics: Experimental Observations and Theoretical Considerationsירדן לויןBelum ada peringkat
- OkokosDokumen3 halamanOkokoskaulauBelum ada peringkat
- Pharmacokinetic: Herni SupraptiDokumen76 halamanPharmacokinetic: Herni SupraptiveniBelum ada peringkat
- Pharmacology- The Molecular Dance: Understanding Drug Interactions: Harmony and Chaos: The Symphony of Drug InteractionsDari EverandPharmacology- The Molecular Dance: Understanding Drug Interactions: Harmony and Chaos: The Symphony of Drug InteractionsBelum ada peringkat
- Presystemic Drug Elimination: Butterworths International Medical Reviews: Clinical Pharmacology and TherapeuticsDari EverandPresystemic Drug Elimination: Butterworths International Medical Reviews: Clinical Pharmacology and TherapeuticsCharles F. GeorgePenilaian: 1 dari 5 bintang1/5 (1)
- Translational ADMET for Drug Therapy: Principles, Methods, and Pharmaceutical ApplicationsDari EverandTranslational ADMET for Drug Therapy: Principles, Methods, and Pharmaceutical ApplicationsPenilaian: 1 dari 5 bintang1/5 (1)
- Fairy Tail - Main ThemeDokumen3 halamanFairy Tail - Main Theme1234chocoBelum ada peringkat
- Mbs Quick Guide: JULY 2020Dokumen2 halamanMbs Quick Guide: JULY 20201234chocoBelum ada peringkat
- Code Geass - StoriesDokumen5 halamanCode Geass - Stories1234chocoBelum ada peringkat
- Blue BirdDokumen7 halamanBlue Bird1234chocoBelum ada peringkat
- Doctor Talk: Communication Practice Role PlaysDokumen6 halamanDoctor Talk: Communication Practice Role Plays1234chocoBelum ada peringkat
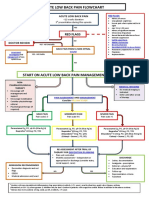
- Acute Low Back Pain Flowchart January 2017Dokumen1 halamanAcute Low Back Pain Flowchart January 20171234chocoBelum ada peringkat
- Topical Steroids (Sep 19) PDFDokumen7 halamanTopical Steroids (Sep 19) PDF1234chocoBelum ada peringkat
- Blue Card Declaration PDFDokumen1 halamanBlue Card Declaration PDF1234chocoBelum ada peringkat
- PBM Module1 MTP Template 0Dokumen2 halamanPBM Module1 MTP Template 01234chocoBelum ada peringkat
- Taking A Social and Cultural HistoryDokumen3 halamanTaking A Social and Cultural History1234chocoBelum ada peringkat
- Managing Mental Illness in Patients From CALD Backgrounds: PsychiatryDokumen5 halamanManaging Mental Illness in Patients From CALD Backgrounds: Psychiatry1234chocoBelum ada peringkat
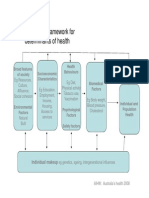
- A Conceptual Framework For HealthDokumen1 halamanA Conceptual Framework For Health1234chocoBelum ada peringkat
- Acute Post-Op Pain ManagementDokumen4 halamanAcute Post-Op Pain Management1234chocoBelum ada peringkat
- Barrett's Esophagus: Identifying The Squamocolumnar (SC) and Gastroesophageal (GE) Junctions EndoscopicallyDokumen10 halamanBarrett's Esophagus: Identifying The Squamocolumnar (SC) and Gastroesophageal (GE) Junctions Endoscopically1234chocoBelum ada peringkat
- Prenatal Screening Test (AUS) PDFDokumen24 halamanPrenatal Screening Test (AUS) PDFshirley_ling_15Belum ada peringkat
- 1 PBDokumen3 halaman1 PBHaola andaniBelum ada peringkat
- Colchicine - General DataDokumen3 halamanColchicine - General DataCatalina BanuBelum ada peringkat
- 1.2.1.1 - Overview of Clinical Trials in Canada - Tanya Ramsamy - eDokumen25 halaman1.2.1.1 - Overview of Clinical Trials in Canada - Tanya Ramsamy - ecristofer BellidoBelum ada peringkat
- ReportDokumen21 halamanReportRenz CruzBelum ada peringkat
- Chemistry Is Important in MedicineDokumen6 halamanChemistry Is Important in MedicineKimberly A. S. Smith0% (1)
- Summary Plan Description Book 1 of 2 Plan U3Dokumen20 halamanSummary Plan Description Book 1 of 2 Plan U3Alfred IDunnoBelum ada peringkat
- (Analytical Profiles of Drug Substances 9) Klaus Florey (Eds.) - Academic Press (1981) PDFDokumen614 halaman(Analytical Profiles of Drug Substances 9) Klaus Florey (Eds.) - Academic Press (1981) PDFAstrid Carolina Gutierrez ShimabukuroBelum ada peringkat
- DESIGN, DEVELOPMENT AND IN-VITRO EVALUATION OF METOPROLOL TARTRATE FAST DISSOLVING TABLETS K. Ramesh Reddy, V. Jayasankar Reddy, G. Saisri Harsha, K.AnilDokumen8 halamanDESIGN, DEVELOPMENT AND IN-VITRO EVALUATION OF METOPROLOL TARTRATE FAST DISSOLVING TABLETS K. Ramesh Reddy, V. Jayasankar Reddy, G. Saisri Harsha, K.AniliajpsBelum ada peringkat
- Drug Metabolism & Pharmacokinetics in Drug Discovery: A Primer For Bioanalytical Chemists, Part IDokumen7 halamanDrug Metabolism & Pharmacokinetics in Drug Discovery: A Primer For Bioanalytical Chemists, Part IJoynalIslamKabirBelum ada peringkat
- DsDokumen9 halamanDsVavaxxxRemusaBelum ada peringkat
- Management of Cardiac ArrestDokumen10 halamanManagement of Cardiac ArrestAhmed VelićBelum ada peringkat
- Zidovudineinduced AnaemiaDokumen23 halamanZidovudineinduced AnaemiaSuresh ThanneruBelum ada peringkat
- Pharma Domain 101 - The Industry Lingo (Latest)Dokumen10 halamanPharma Domain 101 - The Industry Lingo (Latest)Ankit AgarwalBelum ada peringkat
- StorageDokumen2 halamanStorageShagorShagorBelum ada peringkat
- Skripsi KustaDokumen61 halamanSkripsi Kustaandre yuindartantoBelum ada peringkat
- Myasthenia GDokumen7 halamanMyasthenia GAcbel RnucoBelum ada peringkat
- Shaylyn Pady ResumeDokumen2 halamanShaylyn Pady Resumeapi-383927437Belum ada peringkat
- Research Journal of Pharmaceutical, Biological and Chemical SciencesDokumen5 halamanResearch Journal of Pharmaceutical, Biological and Chemical SciencesDessy Erlyani Mugita SariBelum ada peringkat
- Protein Binding of Drug: Page - 1 CMR College of Pharmacy, 2013Dokumen18 halamanProtein Binding of Drug: Page - 1 CMR College of Pharmacy, 2013REDDYGAARI ABBAYIBelum ada peringkat
- Introduction To Pharmaceutical Aerosols: D.D Moyo Pharmaceutical Technology 2011Dokumen34 halamanIntroduction To Pharmaceutical Aerosols: D.D Moyo Pharmaceutical Technology 2011valentine mhuteBelum ada peringkat
- Emulsifier For Personal CareDokumen5 halamanEmulsifier For Personal CareWaltoy DinizBelum ada peringkat
- ASSAY METHOD DEVELOPMENT AND VALIDATION FOR THE ESTIMATION OF SOLIFENACIN SUCCINATE IN TABLETS BY UV SPECTROPHOTOMETRY N.J.R. Hepsebah, A. Ashok KumarDokumen6 halamanASSAY METHOD DEVELOPMENT AND VALIDATION FOR THE ESTIMATION OF SOLIFENACIN SUCCINATE IN TABLETS BY UV SPECTROPHOTOMETRY N.J.R. Hepsebah, A. Ashok KumariajpsBelum ada peringkat
- Cosmetic Chemistry VocabularyDokumen2 halamanCosmetic Chemistry Vocabularyapi-266663713Belum ada peringkat
- Encube Corporate PresentationDokumen19 halamanEncube Corporate PresentationAnnu KambleBelum ada peringkat