Anda mungkin juga menyukai
- Bendall Et Al ScienceDokumen11 halamanBendall Et Al SciencePratip ChattopadhyayBelum ada peringkat
- Anucleate Platelets Generate Progeny 1Dokumen34 halamanAnucleate Platelets Generate Progeny 1Alejandro BarretoBelum ada peringkat
- LAPORAN PRAKTIKUM Apoptosis Moh Jaml 226070103141001Dokumen11 halamanLAPORAN PRAKTIKUM Apoptosis Moh Jaml 226070103141001jimi jamalBelum ada peringkat
- Ancillary Studies in Pleural, Pericardial, and Peritoneal EffusionDokumen9 halamanAncillary Studies in Pleural, Pericardial, and Peritoneal EffusionRoli CuizzBelum ada peringkat
- A Mathematical Dissection of The Adaptation of Cell Populations To Fluctuating Oxygen LevelsDokumen24 halamanA Mathematical Dissection of The Adaptation of Cell Populations To Fluctuating Oxygen LevelsCarolina RibeiroBelum ada peringkat
- Diagnosis of Blood and Bone Marrow DisordersDari EverandDiagnosis of Blood and Bone Marrow DisordersSa A. WangBelum ada peringkat
- Tissue PDFDokumen11 halamanTissue PDFdewiarisandyBelum ada peringkat
- Micromachines 08 00015Dokumen26 halamanMicromachines 08 00015yashbhardwajBelum ada peringkat
- 10 1002@bit 26476Dokumen33 halaman10 1002@bit 26476Jamie SamuelBelum ada peringkat
- Thrombosis and Bleeding Disorders: Theory and MethodsDari EverandThrombosis and Bleeding Disorders: Theory and MethodsNils U. BangPenilaian: 2 dari 5 bintang2/5 (1)
- SelekmanDokumen14 halamanSelekmanguadelupehBelum ada peringkat
- Umbilical Cord Blood Stem Cells - Potential Therapeutic Tool For Neural Injuries and DisordersDokumen9 halamanUmbilical Cord Blood Stem Cells - Potential Therapeutic Tool For Neural Injuries and DisordersIgorpin2Belum ada peringkat
- Immune Cell PhenotypingDokumen24 halamanImmune Cell PhenotypingSavina SabahBelum ada peringkat
- Blood RecreationDokumen382 halamanBlood Recreationhasla17Belum ada peringkat
- Blood RecreationDokumen387 halamanBlood RecreationAlexandr TrotskyBelum ada peringkat
- 06-Cryopreservation of Stem CellsDokumen29 halaman06-Cryopreservation of Stem Cellsjashuj.2004Belum ada peringkat
- Cytometry Part B Clinical - 2003 - Mart Nez - Routine Use of Immunophenotype by Flow Cytometry in Tissues With SuspectedDokumen8 halamanCytometry Part B Clinical - 2003 - Mart Nez - Routine Use of Immunophenotype by Flow Cytometry in Tissues With SuspectedGiorgia RisiBelum ada peringkat
- HS BCSH Sept 2011Dokumen40 halamanHS BCSH Sept 2011lidya_zhuangBelum ada peringkat
- Educational Guide: 2nd EditionDokumen130 halamanEducational Guide: 2nd EditionEdin HamzicBelum ada peringkat
- Erythrocyte Ageing in Vivo and in Vitro - Structural Aspects and Implications For TransfusionDokumen13 halamanErythrocyte Ageing in Vivo and in Vitro - Structural Aspects and Implications For TransfusionPritha BhuwapaksophonBelum ada peringkat
- Metabolic Fingerprints of Human Primary Endothelial and Fibroblast CellsDokumen12 halamanMetabolic Fingerprints of Human Primary Endothelial and Fibroblast CellsDavid Galiano LatorreBelum ada peringkat
- Bone Marrow Structure HistologyDokumen18 halamanBone Marrow Structure HistologyJulissa Juarez CastilloBelum ada peringkat
- Pi Is 0092867419311213Dokumen15 halamanPi Is 0092867419311213skodgeBelum ada peringkat
- Cheuk 2004Dokumen23 halamanCheuk 2004wendy aide castro moraBelum ada peringkat
- PIIS2213671114002951Dokumen15 halamanPIIS2213671114002951Ana LimeiraBelum ada peringkat
- Carraway 2010Dokumen2 halamanCarraway 2010bixagif369Belum ada peringkat
- FlowcytometryDokumen10 halamanFlowcytometryWidia MeliawatiBelum ada peringkat
- Integrated Analysis of Ultra-Deep ProteomesDokumen20 halamanIntegrated Analysis of Ultra-Deep ProteomesMAURICIO FLORESBelum ada peringkat
- Chromosone AnalysisDokumen234 halamanChromosone AnalysisSpataru VasileBelum ada peringkat
- FGS Renal Cell CADokumen14 halamanFGS Renal Cell CAselcukorkmazBelum ada peringkat
- 9 Flow Cytometric Strategies To Study CNS DevelopmentDokumen32 halaman9 Flow Cytometric Strategies To Study CNS DevelopmenttaroBelum ada peringkat
- Cancer Res 2006 Michaud 6463 7Dokumen6 halamanCancer Res 2006 Michaud 6463 7Arul PrakashBelum ada peringkat
- BMM 13 63Dokumen10 halamanBMM 13 63Glauce L TrevisanBelum ada peringkat
- Posudek Oponenta AlisiDokumen5 halamanPosudek Oponenta AlisiAbdulJawad Ibrahim ElmezoghiBelum ada peringkat
- Nanoparticle-Induced Vascular Blockade in Human Prostate CancerDokumen11 halamanNanoparticle-Induced Vascular Blockade in Human Prostate CancerMr-Mohamed AliBelum ada peringkat
- Characterization of CD147, CA9, and CD70 As Tumor-Specific Markers On Extracellular Vesicles in Clear Cell Renal Cell CarcinomaDokumen17 halamanCharacterization of CD147, CA9, and CD70 As Tumor-Specific Markers On Extracellular Vesicles in Clear Cell Renal Cell Carcinomahu ziBelum ada peringkat
- Immunostaining of Voltage-Gated Ion Channels in Cell Lines and Neurons - Key Concepts and Potential PitfallsDokumen27 halamanImmunostaining of Voltage-Gated Ion Channels in Cell Lines and Neurons - Key Concepts and Potential Pitfallskjf185Belum ada peringkat
- DDAKO Flow Cytometry Educational Guide 2nd Ed 2006Dokumen68 halamanDDAKO Flow Cytometry Educational Guide 2nd Ed 2006topcat01Belum ada peringkat
- Insights New111Dokumen13 halamanInsights New111cassidy conchaBelum ada peringkat
- PLOSone2016 PDFDokumen20 halamanPLOSone2016 PDFmfcarBelum ada peringkat
- 2023 - Decoding of Exosome Heterogeneity For Cancer Theranostics - PR - Check RefrencesDokumen3 halaman2023 - Decoding of Exosome Heterogeneity For Cancer Theranostics - PR - Check Refrencesfaez 80Belum ada peringkat
- 1 s2.0 S0168165623000123 MainDokumen10 halaman1 s2.0 S0168165623000123 MainLauren Caluya LañoBelum ada peringkat
- Flow CytometryDokumen7 halamanFlow CytometryMayHnin KhaingBelum ada peringkat
- Multiline Age Potential of Adult Human Mesenchymal Stem CellsDokumen7 halamanMultiline Age Potential of Adult Human Mesenchymal Stem CellsDeepak VelusamyBelum ada peringkat
- Myeloid Cell Origins, Differentiation, and Clinical ImplicationsDokumen18 halamanMyeloid Cell Origins, Differentiation, and Clinical ImplicationsWendy PBelum ada peringkat
- Characterization of Circulating Tumor Cells by Fluorescence in Situ HybridizationDokumen8 halamanCharacterization of Circulating Tumor Cells by Fluorescence in Situ HybridizationNidhi JaisBelum ada peringkat
- Cell Biology Assays: ProteinsDari EverandCell Biology Assays: ProteinsFanny JaulinBelum ada peringkat
- Alsaleh Et Al., 20196 (MS)Dokumen10 halamanAlsaleh Et Al., 20196 (MS)AngelBelum ada peringkat
- Mesenchymal Stem Cells Literature ReviewDokumen9 halamanMesenchymal Stem Cells Literature ReviewafdtygyhkBelum ada peringkat
- 2 A Rapid High-Precision Flow Cytometry Based Technique For Total WhiteDokumen14 halaman2 A Rapid High-Precision Flow Cytometry Based Technique For Total WhitePablo LópezBelum ada peringkat
- Mekanisme Stem CellDokumen17 halamanMekanisme Stem CellKEBIDANAN 16 UNHAS UNHASBelum ada peringkat
- Flow Cytometry-Clinical ApplicationsDokumen5 halamanFlow Cytometry-Clinical ApplicationscandiddreamsBelum ada peringkat
- Basofilos 1Dokumen8 halamanBasofilos 1Natalia Cano GBelum ada peringkat
- Nelson 2015Dokumen8 halamanNelson 2015Thanasis PapatheodorouBelum ada peringkat
- Cancer Stem Cells Problems For TherapyDokumen15 halamanCancer Stem Cells Problems For Therapyapi-162161895Belum ada peringkat
- Immortalized Liver Endothel CCmodelDokumen20 halamanImmortalized Liver Endothel CCmodelAndreiBelum ada peringkat
- Cell Death Assays For Drug DiscoveryDokumen17 halamanCell Death Assays For Drug DiscoveryMariano PerezBelum ada peringkat
- Molecular Analysis To Demonstrate That Odontogenic Keratocysts Are NeoplasticDokumen5 halamanMolecular Analysis To Demonstrate That Odontogenic Keratocysts Are NeoplasticSamar KhanBelum ada peringkat
- AulstDokumen38 halamanAulstAulia Azizah KosmanBelum ada peringkat
- Case Report:: Stemi Anteroseptal Wall OnsetDokumen38 halamanCase Report:: Stemi Anteroseptal Wall OnsetAulia Azizah KosmanBelum ada peringkat
- AnatomiDokumen34 halamanAnatomiAulia Azizah KosmanBelum ada peringkat
- Soal Uro InterDokumen12 halamanSoal Uro InterRaihan Zulfiqar HalimBelum ada peringkat
- Manual - CSL 1 Dasar RadiologiDokumen3 halamanManual - CSL 1 Dasar RadiologiAulia Azizah KosmanBelum ada peringkat
- Table BMIDokumen1 halamanTable BMIAulia Azizah KosmanBelum ada peringkat
- Laporan HidrolikaDokumen42 halamanLaporan HidrolikaAulia Azizah KosmanBelum ada peringkat
- ImunoDokumen11 halamanImunoAulia Azizah KosmanBelum ada peringkat
- Pex 04 01Dokumen7 halamanPex 04 01Aulia Azizah KosmanBelum ada peringkat
- Breastfeeding QuestionnaireDokumen2 halamanBreastfeeding QuestionnaireDr Puteri Nur Sabrina Binti Mohd HanapiBelum ada peringkat
- Medical-Surgical Nursing 1Dokumen28 halamanMedical-Surgical Nursing 1Maui Ting100% (1)
- Sabouraud Dextrose BrothDokumen2 halamanSabouraud Dextrose Brothrdn2111Belum ada peringkat
- Darvocet N Drug CardDokumen1 halamanDarvocet N Drug CardSheri490Belum ada peringkat
- Unpaywall ArticleDokumen2 halamanUnpaywall ArticleChristian HdezBelum ada peringkat
- Annual Drug Data Report Vol-1 1971Dokumen228 halamanAnnual Drug Data Report Vol-1 1971lasicablava50% (2)
- Apraxia of Speech and Grammatical Language Impairment in Children With Autism Procedural Deficit HypothesisDokumen6 halamanApraxia of Speech and Grammatical Language Impairment in Children With Autism Procedural Deficit HypothesisEditor IJTSRDBelum ada peringkat
- RRL LocalinternationalDokumen11 halamanRRL LocalinternationalkdfhjfhfBelum ada peringkat
- L7 ConfoundingDokumen57 halamanL7 ConfoundinggygBelum ada peringkat
- Hidetaka Nomura Et Al. (2015)Dokumen4 halamanHidetaka Nomura Et Al. (2015)Janardana GedeBelum ada peringkat
- Effectiveness of Workplace Counseling On Employee Performance. A Case of Mumias Sugar Company Limited, KenyaDokumen10 halamanEffectiveness of Workplace Counseling On Employee Performance. A Case of Mumias Sugar Company Limited, KenyainventionjournalsBelum ada peringkat
- Imaging of Intracranial InfectionsDokumen12 halamanImaging of Intracranial Infectionsrafael rocha novaesBelum ada peringkat
- Exercise Science Honours 2016Dokumen22 halamanExercise Science Honours 2016asdgffdBelum ada peringkat
- Nursing Care Plan: Student's Name: ID: Course Name: SettingDokumen3 halamanNursing Care Plan: Student's Name: ID: Course Name: SettingnjoodBelum ada peringkat
- Biology Form 4 Chapter 5 Cell DivisionDokumen7 halamanBiology Form 4 Chapter 5 Cell Divisiongelgaban67% (3)
- Bee Empowerment Leslie WilsonDokumen6 halamanBee Empowerment Leslie Wilsonchakravartys1327100% (1)
- Target Product Profile Imbruvica - IbrutinibDokumen1 halamanTarget Product Profile Imbruvica - IbrutinibDiti ShahBelum ada peringkat
- Urticaria and Angioedema: Bettina WediDokumen26 halamanUrticaria and Angioedema: Bettina WediMerlin MuktialiBelum ada peringkat
- ENT Atlas Web PDFDokumen442 halamanENT Atlas Web PDFIsabella CrăciunBelum ada peringkat
- Fat Loss For LifeDokumen42 halamanFat Loss For LifeDjuli Ona83% (12)
- Scheme of Instruction IIScDokumen236 halamanScheme of Instruction IIScRS1678Belum ada peringkat
- 04-15-21 Covid-19 Vaccine TrackerDokumen52 halaman04-15-21 Covid-19 Vaccine TrackerJohn Michael JetiganBelum ada peringkat
- Anemia Differential Diagnosis : Microcytic Normocytic MacrocyticDokumen1 halamanAnemia Differential Diagnosis : Microcytic Normocytic Macrocyticمحمد عقيلي100% (1)
- Biochemistry of Jaundice 03Dokumen52 halamanBiochemistry of Jaundice 03Cathleen May Dela Cruz50% (2)
- Course Task 19Dokumen1 halamanCourse Task 19Laira CañeteBelum ada peringkat
- Copd Action Plan PDFDokumen2 halamanCopd Action Plan PDFdtech2Belum ada peringkat
- Your Body Believes Every Word Y - Barbara Hoberman LevineDokumen380 halamanYour Body Believes Every Word Y - Barbara Hoberman LevineRoryBradshawBelum ada peringkat
- Immunology MCQ BY: Dr. AlgassimDokumen4 halamanImmunology MCQ BY: Dr. AlgassimmohamedBelum ada peringkat
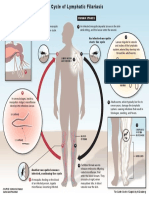
- Lymphatic Filariasis Life Cycle PDFDokumen1 halamanLymphatic Filariasis Life Cycle PDFlauraBelum ada peringkat
- Rhinitis MedicamentosaDokumen22 halamanRhinitis MedicamentosaDanti NurdindayantiBelum ada peringkat