Anda mungkin juga menyukai
- Terming Prime MinisterDokumen1 halamanTerming Prime MinisterbenaminsanBelum ada peringkat
- Your Mind Is Your LimitationDokumen4 halamanYour Mind Is Your LimitationbenaminsanBelum ada peringkat
- Your Mind Is Your Own LimitationDokumen3 halamanYour Mind Is Your Own LimitationbenaminsanBelum ada peringkat
- Presentation 4Dokumen31 halamanPresentation 4benaminsanBelum ada peringkat
- Dsa MCQDokumen9 halamanDsa MCQbenaminsan100% (1)
- Quiz 3 - AnsDokumen3 halamanQuiz 3 - AnsRahul AroraBelum ada peringkat
- Scanned 20160529-2301Dokumen1 halamanScanned 20160529-2301benaminsanBelum ada peringkat
- WordsDokumen2 halamanWordsbenaminsanBelum ada peringkat
- DeadpoolDokumen1 halamanDeadpoolbenaminsanBelum ada peringkat
- German Tutorials Basic PhrasesDokumen200 halamanGerman Tutorials Basic Phrasesbenaminsan100% (1)
- ViewDokumen2 halamanViewbenaminsanBelum ada peringkat
- C Lecture NotesDokumen188 halamanC Lecture Notessurendraaalla0% (1)
- TestDokumen1 halamanTestbenaminsanBelum ada peringkat
- Flyweight Pattern: HistoryDokumen3 halamanFlyweight Pattern: HistorybenaminsanBelum ada peringkat
- A Pattern Matching Approach For Clustering Gene Expression DataDokumen20 halamanA Pattern Matching Approach For Clustering Gene Expression DatabenaminsanBelum ada peringkat
- Multiple Choice Quiz-1Dokumen4 halamanMultiple Choice Quiz-1benaminsanBelum ada peringkat
- Building Vocabulary and Language SkillsDokumen1 halamanBuilding Vocabulary and Language SkillsbenaminsanBelum ada peringkat
- Consuming OData Service in JavaScriptDokumen19 halamanConsuming OData Service in JavaScriptmohammedmeeran3061Belum ada peringkat
- CNIS Preparing For An InterviewDokumen5 halamanCNIS Preparing For An InterviewEnryu NagantBelum ada peringkat
- CommandsDokumen1 halamanCommandsbenaminsanBelum ada peringkat
- OracleDokumen1 halamanOraclebenaminsanBelum ada peringkat
- GATE 2014: Syllabus For Computer Science and Information Technology (CS)Dokumen4 halamanGATE 2014: Syllabus For Computer Science and Information Technology (CS)Prathmesh KalaBelum ada peringkat
- UGProject GuidelinesDokumen16 halamanUGProject GuidelinesbenaminsanBelum ada peringkat
- Resume Writing and Job Search Correspondence: Career Development OfficeDokumen37 halamanResume Writing and Job Search Correspondence: Career Development Officebenaminsan100% (1)
- Comp322 Lec2 f09 v1 PDFDokumen25 halamanComp322 Lec2 f09 v1 PDFbenaminsanBelum ada peringkat
- Computer Networks MCQDokumen8 halamanComputer Networks MCQbenaminsan100% (4)
- Pattern MatchingDokumen58 halamanPattern Matchingamitavaghosh1Belum ada peringkat
- HCL 1Dokumen8 halamanHCL 1anil khannaBelum ada peringkat
- Cisco pp2Dokumen5 halamanCisco pp2benaminsanBelum ada peringkat
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5784)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (890)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (399)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (72)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (119)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- Wegsheider's Role Theory in Substance Abusing FamiliesDokumen5 halamanWegsheider's Role Theory in Substance Abusing FamiliesLouisa KouklaBelum ada peringkat
- Papal InfallibilityDokumen6 halamanPapal InfallibilityFrancis AkalazuBelum ada peringkat
- Advanced Management Accounting: ConceptsDokumen47 halamanAdvanced Management Accounting: ConceptsGEDDIGI BHASKARREDDYBelum ada peringkat
- Geographic Location SystemsDokumen16 halamanGeographic Location SystemsSyed Jabed Miadad AliBelum ada peringkat
- Petitioner Respondent: Civil Service Commission, - Engr. Ali P. DaranginaDokumen4 halamanPetitioner Respondent: Civil Service Commission, - Engr. Ali P. Daranginaanika fierroBelum ada peringkat
- Sound of SundayDokumen3 halamanSound of SundayJean PaladanBelum ada peringkat
- Department of Information TechnologyDokumen1 halamanDepartment of Information TechnologyMuhammad ZeerakBelum ada peringkat
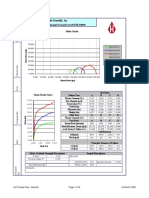
- Humboldt Scientific, Inc.: Normal Stress (Psi)Dokumen9 halamanHumboldt Scientific, Inc.: Normal Stress (Psi)Dilson Loaiza CruzBelum ada peringkat
- Copticmanuscript 00 CoelDokumen468 halamanCopticmanuscript 00 Coelbavly barsomBelum ada peringkat
- ABC Pre School: (Please Refer Advertisement in This Section)Dokumen5 halamanABC Pre School: (Please Refer Advertisement in This Section)hemacrcBelum ada peringkat
- Comparative Summary of 5 Learning TheoriesDokumen2 halamanComparative Summary of 5 Learning TheoriesMonique Gabrielle Nacianceno AalaBelum ada peringkat
- Commonwealth scholarships to strengthen global health systemsDokumen4 halamanCommonwealth scholarships to strengthen global health systemsanonymous machineBelum ada peringkat
- Person On The StreetDokumen12 halamanPerson On The StreetalyssajdorazioBelum ada peringkat
- EMship Course ContentDokumen82 halamanEMship Course ContentBecirspahic Almir100% (1)
- Quieting of TitleDokumen11 halamanQuieting of TitleJONA PHOEBE MANGALINDANBelum ada peringkat
- M I Ngày M T Trò Chơi PDFDokumen354 halamanM I Ngày M T Trò Chơi PDFkaka_02468100% (1)
- List of TNA MaterialsDokumen166 halamanList of TNA MaterialsRamesh NayakBelum ada peringkat
- Signals SyllabusDokumen1 halamanSignals SyllabusproBelum ada peringkat
- Certification 5Dokumen10 halamanCertification 5juliet.clementeBelum ada peringkat
- 2nd Year Pak Studies (English Medium)Dokumen134 halaman2nd Year Pak Studies (English Medium)MUHAMMAD HASAN RAHIMBelum ada peringkat
- BA 424 Chapter 1 NotesDokumen6 halamanBA 424 Chapter 1 Notesel jiBelum ada peringkat
- Sector:: Automotive/Land Transport SectorDokumen20 halamanSector:: Automotive/Land Transport SectorVedin Padilla Pedroso92% (12)
- Ahmed Bahri Omar 1Dokumen7 halamanAhmed Bahri Omar 1l3gsdBelum ada peringkat
- Pan or The DevilDokumen8 halamanPan or The DevilMarguerite and Leni Johnson100% (1)
- Call HandlingDokumen265 halamanCall HandlingABHILASHBelum ada peringkat
- BSIA - Access ControlDokumen16 halamanBSIA - Access ControlSayed HashemBelum ada peringkat
- List of As... SimilesDokumen3 halamanList of As... SimilesFara ZahariBelum ada peringkat
- Chain of Custody Requirement in Drug CasesDokumen23 halamanChain of Custody Requirement in Drug CasesMiw CortesBelum ada peringkat
- Quantum Mechanics and PeriodicityDokumen51 halamanQuantum Mechanics and Periodicitynxumalopat2Belum ada peringkat
- Group 2Dokumen3 halamanGroup 2sharmisthahalder21Belum ada peringkat