Anda mungkin juga menyukai
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (119)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (399)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (587)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2219)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- Science in Action 9 PDFDokumen542 halamanScience in Action 9 PDFCalvin Thomas47% (15)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (890)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- Know Your Magnetic FieldDokumen75 halamanKnow Your Magnetic FieldAtma Jnani100% (5)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- Stage Lighting Technician Ebook PDFDokumen207 halamanStage Lighting Technician Ebook PDFJoao Kodama100% (2)
- Moulded Case Circuit Breakers Technical OverviewDokumen0 halamanMoulded Case Circuit Breakers Technical OverviewJoni Efwan100% (2)
- VIVA Questions - WPCDokumen18 halamanVIVA Questions - WPCvinayBelum ada peringkat
- Physics For IIT JEE Vol 2 How To Read The BookDokumen24 halamanPhysics For IIT JEE Vol 2 How To Read The BookNaveen Kumar SinghBelum ada peringkat
- Wa0002Dokumen164 halamanWa0002wilson simfukweBelum ada peringkat
- Battery HandbookDokumen16 halamanBattery Handbookprakdora100% (1)
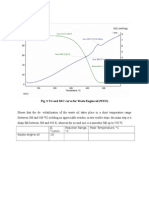
- Fig. 3 TG and DSC Curve For Waste Engine Oil (WEO)Dokumen1 halamanFig. 3 TG and DSC Curve For Waste Engine Oil (WEO)sumit rajBelum ada peringkat
- re-HS 514Dokumen9 halamanre-HS 514sumit rajBelum ada peringkat
- AnwarDokumen17 halamanAnwarsumit rajBelum ada peringkat
- List Components For PurchaseDokumen5 halamanList Components For Purchasesumit rajBelum ada peringkat
- Static Watt MeterDokumen6 halamanStatic Watt MeterChandra Bose KnBelum ada peringkat
- Choosing An AC or DC Coil For A Solenoid Valve - Tameson PDFDokumen4 halamanChoosing An AC or DC Coil For A Solenoid Valve - Tameson PDFeakonakosBelum ada peringkat
- Necp-How To Select Brushes For Motors and Generators2 PDFDokumen16 halamanNecp-How To Select Brushes For Motors and Generators2 PDFJason CarrBelum ada peringkat
- Led Characteristics ManualDokumen3 halamanLed Characteristics ManualRoxxxBelum ada peringkat
- 0653 s16 QP 61Dokumen20 halaman0653 s16 QP 61yuke kristinaBelum ada peringkat
- ACS752 100 DatasheetDokumen9 halamanACS752 100 DatasheetKayro PereiraBelum ada peringkat
- Theory and Physics of Abrams Electronic ReactionDokumen7 halamanTheory and Physics of Abrams Electronic ReactionaaaaBelum ada peringkat
- A. Basic ConceptsDokumen40 halamanA. Basic ConceptsBarkley VelezBelum ada peringkat
- Nagar 1985Dokumen11 halamanNagar 1985drjonesg19585102Belum ada peringkat
- Signal Recovery From Noise in Electronic Instrumentation NOE0750300582Dokumen238 halamanSignal Recovery From Noise in Electronic Instrumentation NOE0750300582ssamonasBelum ada peringkat
- AC Corrosion of Metals ResearchDokumen168 halamanAC Corrosion of Metals ResearchMn Hjhjj100% (1)
- INDUCTANCE OF AN INDUCTOR EXPLAINEDDokumen36 halamanINDUCTANCE OF AN INDUCTOR EXPLAINEDDanelBelum ada peringkat
- Physics ProjectDokumen13 halamanPhysics Projectdj gangsterBelum ada peringkat
- Lemon BatteryDokumen7 halamanLemon BatteryVijay GbBelum ada peringkat
- ABB Voltage Limiting Device HVL 120-0.3 - Data Sheet 1HC0075863 AE ENDokumen6 halamanABB Voltage Limiting Device HVL 120-0.3 - Data Sheet 1HC0075863 AE ENn srikanthBelum ada peringkat
- Laporan Praktikum Proses Manufaktur 2 Kelompok S25 Modul PM2-01 Shift Sabtu Siang PDFDokumen30 halamanLaporan Praktikum Proses Manufaktur 2 Kelompok S25 Modul PM2-01 Shift Sabtu Siang PDFGilang Tito F.Belum ada peringkat
- ICAT Syllbus For Part 66Dokumen28 halamanICAT Syllbus For Part 66AzriSafwanRusliBelum ada peringkat
- Project 3 Technical NDT DocumentDokumen18 halamanProject 3 Technical NDT Documentapi-242490471Belum ada peringkat
- Smart Water Quality Monitoring System For Real Time ApplicationsDokumen8 halamanSmart Water Quality Monitoring System For Real Time ApplicationsMaruti SankannanavarBelum ada peringkat
- To Determine The Internal Resistance of A Given Primary Cell Using Potentiometer - Learn CBSEDokumen11 halamanTo Determine The Internal Resistance of A Given Primary Cell Using Potentiometer - Learn CBSEAbhay Pratap GangwarBelum ada peringkat
- Chapter 8: Band Theory of Solids Concept of Free Electron Theory: Hour 1Dokumen25 halamanChapter 8: Band Theory of Solids Concept of Free Electron Theory: Hour 1Vivek kapoorBelum ada peringkat
- Summary Notes - Topic 2 ElectricityDokumen6 halamanSummary Notes - Topic 2 ElectricitydilsharakaviBelum ada peringkat