Anda mungkin juga menyukai
- The Equation of State For Gases and Liquids: Nobel Lecture, December 12, 1910Dokumen12 halamanThe Equation of State For Gases and Liquids: Nobel Lecture, December 12, 1910AMIT PRAJAPATIBelum ada peringkat
- The Equation of State For Gases and Liquids: Nobel Lecture, December 12, 1910Dokumen12 halamanThe Equation of State For Gases and Liquids: Nobel Lecture, December 12, 1910Nataly CGBelum ada peringkat
- De Broglie WavelengthDokumen3 halamanDe Broglie WavelengthMarion Alyssa100% (1)
- ArticleDokumen9 halamanArticleStella MaryBelum ada peringkat
- GG Stokes 1845 PDFDokumen55 halamanGG Stokes 1845 PDFAnkit soniBelum ada peringkat
- Van Der Waals EquationDokumen10 halamanVan Der Waals EquationBraulio CasanuevaBelum ada peringkat
- Inversion EquationDokumen4 halamanInversion EquationAdil ShaffeeBelum ada peringkat
- Real Gases ArticlesDokumen15 halamanReal Gases ArticlesHabibBelum ada peringkat
- Fundamentals of Chemistry Course Code: ENV-1105Dokumen101 halamanFundamentals of Chemistry Course Code: ENV-1105Mashrufa HussainBelum ada peringkat
- Investigations On The Theory of The Photographic Process - Sheppard and MeesDokumen366 halamanInvestigations On The Theory of The Photographic Process - Sheppard and MeesJorgeCamachoBelum ada peringkat
- A. Chamblin and R. Emparan - Bubbles in Kaluza-Klein Theories With Spacelike or Timelike Internal DimensionsDokumen12 halamanA. Chamblin and R. Emparan - Bubbles in Kaluza-Klein Theories With Spacelike or Timelike Internal DimensionsHuntsmithBelum ada peringkat
- Chemistry 9 NotesDokumen4 halamanChemistry 9 NotesMuhammad AsifBelum ada peringkat
- Chapter Review 3Dokumen4 halamanChapter Review 3Othello McGinnisBelum ada peringkat
- Satish Chandra: Unit - I, Real GasesDokumen6 halamanSatish Chandra: Unit - I, Real GasesSanchari BiswasBelum ada peringkat
- EOS Van - Der - Waals - Equation Nov 2 2011Dokumen7 halamanEOS Van - Der - Waals - Equation Nov 2 2011Gilvan PirôpoBelum ada peringkat
- Equation of State - WikipediaDokumen78 halamanEquation of State - WikipediaPrecious OluwadahunsiBelum ada peringkat
- Bubbling Kun NiDokumen7 halamanBubbling Kun NiBelen CRBelum ada peringkat
- Igcse 52 Solidsliquids&GasesDokumen39 halamanIgcse 52 Solidsliquids&GasesHany ElGezawy100% (1)
- Properties of Liquids and Intermolecular Forces: General Chemistry 2Dokumen53 halamanProperties of Liquids and Intermolecular Forces: General Chemistry 2Cielo GatdulaBelum ada peringkat
- Time and Background Independence: A ResponseDokumen7 halamanTime and Background Independence: A ResponsecalamartBelum ada peringkat
- General Chemistry 2 - q3 - Slm2Dokumen10 halamanGeneral Chemistry 2 - q3 - Slm2Jonnel RoqueBelum ada peringkat
- SS1 Chemistry 2nd Term Lesson Note PDFDokumen58 halamanSS1 Chemistry 2nd Term Lesson Note PDFKelly Isaac100% (3)
- Properties of MatterDokumen8 halamanProperties of MatterAbdur RahmanBelum ada peringkat
- Physical PharmacyDokumen190 halamanPhysical PharmacyKate EvangelistaBelum ada peringkat
- Thermal Physics NotesDokumen17 halamanThermal Physics NotesNo PainBelum ada peringkat
- Pit Zer 1981Dokumen8 halamanPit Zer 1981Niraj ThakreBelum ada peringkat
- LMS TEMPLATE - NOTES Chap 4Dokumen6 halamanLMS TEMPLATE - NOTES Chap 4AlexBelum ada peringkat
- Can Thought Experiments Be Resolved by Experiment? The Case of Aristotle's WheelDokumen20 halamanCan Thought Experiments Be Resolved by Experiment? The Case of Aristotle's WheelItsmeBelum ada peringkat
- A Statistical Theory of Monomolecular Reactions PDFDokumen9 halamanA Statistical Theory of Monomolecular Reactions PDFwenhuishenBelum ada peringkat
- Kinetic Theory (The Gas Laws) - Chemistry Unit IDokumen6 halamanKinetic Theory (The Gas Laws) - Chemistry Unit Imcleodtravis14Belum ada peringkat
- Physica: Viscosity of Carbon Dioxide in The Liquid PhaseDokumen32 halamanPhysica: Viscosity of Carbon Dioxide in The Liquid PhasejtorerocBelum ada peringkat
- G10 SCI Q4WK1 LAS1-checked-and-editedDokumen5 halamanG10 SCI Q4WK1 LAS1-checked-and-editedKirsten Tiffany TriasBelum ada peringkat
- 8 Interpretation Theory of Variables.: Suggested of The Quantum in Tel@asDokumen14 halaman8 Interpretation Theory of Variables.: Suggested of The Quantum in Tel@asSaad KhalidBelum ada peringkat
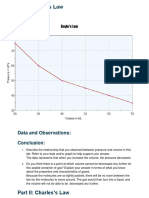
- Data and Observations: ConclusionDokumen2 halamanData and Observations: ConclusionStephany LeviBelum ada peringkat
- Materi 04 - Kimia Teknik - by Waluyo Nuswantoro 2021Dokumen11 halamanMateri 04 - Kimia Teknik - by Waluyo Nuswantoro 2021Ryan BageurBelum ada peringkat
- The History of EntropyDokumen32 halamanThe History of EntropyTiago MartinelliBelum ada peringkat
- 40 Chemistry1702951908Dokumen62 halaman40 Chemistry1702951908ilegbedionkBelum ada peringkat
- Science10 Q4 Mod1 BoyleslawDokumen17 halamanScience10 Q4 Mod1 BoyleslawPrincess PanulayaBelum ada peringkat
- Cadeliña - J - Output Week 1 B in General Chemistry 2Dokumen2 halamanCadeliña - J - Output Week 1 B in General Chemistry 2Jordano CadeliñaBelum ada peringkat
- 1923 Nature V111 p532-233Dokumen3 halaman1923 Nature V111 p532-233Ellena MaggyvinBelum ada peringkat
- Science 10 Las 4-1Dokumen5 halamanScience 10 Las 4-1Michael TuyayBelum ada peringkat
- POGIL: Kinetic Molecular Theory: Learning ObjectivesDokumen3 halamanPOGIL: Kinetic Molecular Theory: Learning ObjectivesSir JoshBelum ada peringkat
- Unsolved Problems in Fluid MechanicsDokumen11 halamanUnsolved Problems in Fluid MechanicsSameh AhmedBelum ada peringkat
- Equation of State: From Wikipedia, The Free EncyclopediaDokumen11 halamanEquation of State: From Wikipedia, The Free EncyclopediaPatricia de LeonBelum ada peringkat
- HW2 WordDokumen3 halamanHW2 WordRudiele SchankoskiBelum ada peringkat
- M2 For Printing PDFDokumen24 halamanM2 For Printing PDFcute chubbitBelum ada peringkat
- Q4 Module 1 CompressedDokumen2 halamanQ4 Module 1 CompressedFELIX ROBERT VALENZUELABelum ada peringkat
- Micelles, Vesicles and Micro EmulsionsDokumen29 halamanMicelles, Vesicles and Micro EmulsionsBárbara Damasceno BragaBelum ada peringkat
- Physical Science - Week 25Dokumen7 halamanPhysical Science - Week 25Mira VeranoBelum ada peringkat
- Entropy: Gibbs' Paradox and The Definition of EntropyDokumen4 halamanEntropy: Gibbs' Paradox and The Definition of Entropynoro70Belum ada peringkat
- Chem. LabDokumen5 halamanChem. LabCel AyunayunBelum ada peringkat
- CEGG2Dokumen26 halamanCEGG2miletinhoBelum ada peringkat
- Resource Guide: - Ideal Gases/Acid Rain & The Greenhouse EffectDokumen22 halamanResource Guide: - Ideal Gases/Acid Rain & The Greenhouse Effectkatherine_strangosBelum ada peringkat
- General Chemistry 1 Week 5 6Dokumen10 halamanGeneral Chemistry 1 Week 5 6Emmanuel ValenzuelaBelum ada peringkat
- Lesson Plan About Gay Lussacs Law 1Dokumen10 halamanLesson Plan About Gay Lussacs Law 1Lyca Mae De Villa100% (2)
- 1407 3162 PDFDokumen97 halaman1407 3162 PDFAnonymous 80p9OVBelum ada peringkat
- The Three States of MatterDokumen25 halamanThe Three States of MatterJelani GreerBelum ada peringkat
- 101.12 IMF Fa19Dokumen6 halaman101.12 IMF Fa19Jossmyr NarceBelum ada peringkat
- Gaussian Process TutorialDokumen59 halamanGaussian Process TutorialEdgar SanchezBelum ada peringkat
- BCI DYN PuRIPD V01 WS1617 Intro General Intro To ModelingDokumen54 halamanBCI DYN PuRIPD V01 WS1617 Intro General Intro To ModelingEdgar SanchezBelum ada peringkat
- Aspen User Guide 10Dokumen936 halamanAspen User Guide 10tryinghard18Belum ada peringkat
- Problem Statement: Schematic For Flow Along A ChannelDokumen9 halamanProblem Statement: Schematic For Flow Along A ChannelEdgar SanchezBelum ada peringkat
- Advanced Process DesignDokumen3 halamanAdvanced Process DesignEdgar SanchezBelum ada peringkat
- Basic EquationsDokumen19 halamanBasic EquationsEdgar SanchezBelum ada peringkat
- InfluenceOfViscous&BouyancyForcesOnMobilzOfResidualTCE PennellEtal EST30 (1996)Dokumen8 halamanInfluenceOfViscous&BouyancyForcesOnMobilzOfResidualTCE PennellEtal EST30 (1996)Edgar SanchezBelum ada peringkat
- Levenspiel 1999 - Chemical Reaction EngineeringDokumen4 halamanLevenspiel 1999 - Chemical Reaction EngineeringDrudervenBelum ada peringkat
- Indoor EcosystemsDokumen3 halamanIndoor EcosystemsEdgar SanchezBelum ada peringkat
- Rayner-Canham 5e Appendix 1 - Thermodynamic Properties of Some Selected Inorganic CompoundsDokumen12 halamanRayner-Canham 5e Appendix 1 - Thermodynamic Properties of Some Selected Inorganic CompoundsEdgar SanchezBelum ada peringkat
- Nucleosintesis 2Dokumen8 halamanNucleosintesis 2Edgar SanchezBelum ada peringkat
- Electric Vehicle Charging Station BplanDokumen19 halamanElectric Vehicle Charging Station BplanAjithBelum ada peringkat
- Zoomlion Gulf FZE Introduction: 1.1 ME Service Support 1.2 Construction CasesDokumen13 halamanZoomlion Gulf FZE Introduction: 1.1 ME Service Support 1.2 Construction CasesArk TradingBelum ada peringkat
- Antonov 225 - The Largest - Airliner in The WorldDokumen63 halamanAntonov 225 - The Largest - Airliner in The WorldFridayFunStuffBelum ada peringkat
- MY-SDK-10000-EE-005 - Method Statement For Concrete Pole Installation - GVB Rev1Dokumen7 halamanMY-SDK-10000-EE-005 - Method Statement For Concrete Pole Installation - GVB Rev1Seeths NairBelum ada peringkat
- Faithful Love: Guitar SoloDokumen3 halamanFaithful Love: Guitar SoloCarol Goldburg33% (3)
- PhysioEx Exercise 1 Activity 1Dokumen3 halamanPhysioEx Exercise 1 Activity 1edvin merida proBelum ada peringkat
- AXIOM75 50 25 1B - Rev.6 10.000MHzDokumen4 halamanAXIOM75 50 25 1B - Rev.6 10.000MHzTürkay PektürkBelum ada peringkat
- Chapter 1Dokumen20 halamanChapter 1Li YuBelum ada peringkat
- Steinecker Boreas: Wort Stripping of The New GenerationDokumen16 halamanSteinecker Boreas: Wort Stripping of The New GenerationAlejandro Javier Delgado AraujoBelum ada peringkat
- Strategi Meningkatkan Kapasitas Penangkar Benih Padi Sawah (Oriza Sativa L) Dengan Optimalisasi Peran Kelompok TaniDokumen24 halamanStrategi Meningkatkan Kapasitas Penangkar Benih Padi Sawah (Oriza Sativa L) Dengan Optimalisasi Peran Kelompok TaniHilmyTafantoBelum ada peringkat
- Omegas Prezentacija 01Dokumen20 halamanOmegas Prezentacija 01Predrag Djordjevic100% (1)
- Where Business Happens Where Happens: SupportDokumen19 halamanWhere Business Happens Where Happens: SupportRahul RamtekkarBelum ada peringkat
- DWDMDokumen41 halamanDWDMKarthik KompelliBelum ada peringkat
- 04 - Crystallogaphy III Miller Indices-Faces-Forms-EditedDokumen63 halaman04 - Crystallogaphy III Miller Indices-Faces-Forms-EditedMaisha MujibBelum ada peringkat
- OD - SAP Connector UtilityDokumen22 halamanOD - SAP Connector UtilityShivani SharmaBelum ada peringkat
- Comparativa Microplex F40 Printronix P8220 enDokumen1 halamanComparativa Microplex F40 Printronix P8220 enangel ricaBelum ada peringkat
- Merchant Accounts Are Bank Accounts That Allow Your Business To Accept Card Payments From CustomersDokumen43 halamanMerchant Accounts Are Bank Accounts That Allow Your Business To Accept Card Payments From CustomersRohit Kumar Baghel100% (1)
- Python PyDokumen19 halamanPython Pyakhilesh kr bhagatBelum ada peringkat
- A Study On Awareness of Mutual Funds and Perception of Investors 2Dokumen89 halamanA Study On Awareness of Mutual Funds and Perception of Investors 2Yashaswini BangeraBelum ada peringkat
- E&i QC Inspector Resum and DocumentsDokumen24 halamanE&i QC Inspector Resum and DocumentsIrfan 786pakBelum ada peringkat
- Trading Book - AGDokumen7 halamanTrading Book - AGAnilkumarGopinathanNairBelum ada peringkat
- Put Them Into A Big Bowl. Serve The Salad in Small Bowls. Squeeze Some Lemon Juice. Cut The Fruits Into Small Pieces. Wash The Fruits. Mix The FruitsDokumen2 halamanPut Them Into A Big Bowl. Serve The Salad in Small Bowls. Squeeze Some Lemon Juice. Cut The Fruits Into Small Pieces. Wash The Fruits. Mix The FruitsNithya SweetieBelum ada peringkat
- Tours and Travel MNGTDokumen16 halamanTours and Travel MNGTArpita Jaiswal100% (5)
- First Certificate Star SB PDFDokumen239 halamanFirst Certificate Star SB PDFPatricia Gallego GálvezBelum ada peringkat
- Nodal Mesh AnalysisDokumen20 halamanNodal Mesh Analysisjaspreet964Belum ada peringkat
- 1.2.2.5 Packet Tracer - Connecting Devices To Build IoTDokumen4 halaman1.2.2.5 Packet Tracer - Connecting Devices To Build IoTyayasan dharamabharataBelum ada peringkat
- France 10-Day ItineraryDokumen3 halamanFrance 10-Day ItineraryYou goabroadBelum ada peringkat
- Chiba International, IncDokumen15 halamanChiba International, IncMiklós SzerdahelyiBelum ada peringkat
- A Modified Linear Programming Method For Distribution System ReconfigurationDokumen6 halamanA Modified Linear Programming Method For Distribution System Reconfigurationapi-3697505Belum ada peringkat
- User Manual - Wellwash ACDokumen99 halamanUser Manual - Wellwash ACAlexandrBelum ada peringkat
- Higgs Discovery: The Power of Empty SpaceDari EverandHiggs Discovery: The Power of Empty SpacePenilaian: 3 dari 5 bintang3/5 (30)
- A Brief History of Time: From the Big Bang to Black HolesDari EverandA Brief History of Time: From the Big Bang to Black HolesPenilaian: 4 dari 5 bintang4/5 (2193)
- A Beginner's Guide to Constructing the Universe: The Mathematical Archetypes of Nature, Art, and ScienceDari EverandA Beginner's Guide to Constructing the Universe: The Mathematical Archetypes of Nature, Art, and SciencePenilaian: 4 dari 5 bintang4/5 (51)
- Dark Matter and the Dinosaurs: The Astounding Interconnectedness of the UniverseDari EverandDark Matter and the Dinosaurs: The Astounding Interconnectedness of the UniversePenilaian: 3.5 dari 5 bintang3.5/5 (69)
- The Simulated Multiverse: An MIT Computer Scientist Explores Parallel Universes, The Simulation Hypothesis, Quantum Computing and the Mandela EffectDari EverandThe Simulated Multiverse: An MIT Computer Scientist Explores Parallel Universes, The Simulation Hypothesis, Quantum Computing and the Mandela EffectPenilaian: 4.5 dari 5 bintang4.5/5 (20)
- Quantum Spirituality: Science, Gnostic Mysticism, and Connecting with Source ConsciousnessDari EverandQuantum Spirituality: Science, Gnostic Mysticism, and Connecting with Source ConsciousnessPenilaian: 4 dari 5 bintang4/5 (6)
- Summary and Interpretation of Reality TransurfingDari EverandSummary and Interpretation of Reality TransurfingPenilaian: 5 dari 5 bintang5/5 (5)
- The Holographic Universe: The Revolutionary Theory of RealityDari EverandThe Holographic Universe: The Revolutionary Theory of RealityPenilaian: 4.5 dari 5 bintang4.5/5 (76)
- Giza: The Tesla Connection: Acoustical Science and the Harvesting of Clean EnergyDari EverandGiza: The Tesla Connection: Acoustical Science and the Harvesting of Clean EnergyBelum ada peringkat
- Knocking on Heaven's Door: How Physics and Scientific Thinking Illuminate the Universe and the Modern WorldDari EverandKnocking on Heaven's Door: How Physics and Scientific Thinking Illuminate the Universe and the Modern WorldPenilaian: 3.5 dari 5 bintang3.5/5 (64)
- Packing for Mars: The Curious Science of Life in the VoidDari EverandPacking for Mars: The Curious Science of Life in the VoidPenilaian: 4 dari 5 bintang4/5 (1395)
- Midnight in Chernobyl: The Story of the World's Greatest Nuclear DisasterDari EverandMidnight in Chernobyl: The Story of the World's Greatest Nuclear DisasterPenilaian: 4.5 dari 5 bintang4.5/5 (410)
- Let There Be Light: Physics, Philosophy & the Dimensional Structure of ConsciousnessDari EverandLet There Be Light: Physics, Philosophy & the Dimensional Structure of ConsciousnessPenilaian: 4.5 dari 5 bintang4.5/5 (57)
- The Tao of Physics: An Exploration of the Parallels between Modern Physics and Eastern MysticismDari EverandThe Tao of Physics: An Exploration of the Parallels between Modern Physics and Eastern MysticismPenilaian: 4 dari 5 bintang4/5 (500)
- Chernobyl 01:23:40: The Incredible True Story of the World's Worst Nuclear DisasterDari EverandChernobyl 01:23:40: The Incredible True Story of the World's Worst Nuclear DisasterPenilaian: 4 dari 5 bintang4/5 (264)
- Chasing Heisenberg: The Race for the Atom BombDari EverandChasing Heisenberg: The Race for the Atom BombPenilaian: 4.5 dari 5 bintang4.5/5 (8)
- The Beginning of Infinity: Explanations That Transform the WorldDari EverandThe Beginning of Infinity: Explanations That Transform the WorldPenilaian: 5 dari 5 bintang5/5 (60)
- Strange Angel: The Otherworldly Life of Rocket Scientist John Whiteside ParsonsDari EverandStrange Angel: The Otherworldly Life of Rocket Scientist John Whiteside ParsonsPenilaian: 4 dari 5 bintang4/5 (94)
- Quantum Physics: What Everyone Needs to KnowDari EverandQuantum Physics: What Everyone Needs to KnowPenilaian: 4.5 dari 5 bintang4.5/5 (49)
- Too Big for a Single Mind: How the Greatest Generation of Physicists Uncovered the Quantum WorldDari EverandToo Big for a Single Mind: How the Greatest Generation of Physicists Uncovered the Quantum WorldPenilaian: 4.5 dari 5 bintang4.5/5 (8)
- Why Time Flies: A Mostly Scientific InvestigationDari EverandWhy Time Flies: A Mostly Scientific InvestigationPenilaian: 3.5 dari 5 bintang3.5/5 (17)
- Vibration and Frequency: How to Get What You Want in LifeDari EverandVibration and Frequency: How to Get What You Want in LifePenilaian: 4.5 dari 5 bintang4.5/5 (13)
- The End of Everything: (Astrophysically Speaking)Dari EverandThe End of Everything: (Astrophysically Speaking)Penilaian: 4.5 dari 5 bintang4.5/5 (157)