Anda mungkin juga menyukai
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- Jurnal TeratoDokumen5 halamanJurnal TeratoSisQha LuCiiajjaBelum ada peringkat
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- Fentanyl inDokumen10 halamanFentanyl inSisQha LuCiiajjaBelum ada peringkat
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- 5045Dokumen8 halaman5045SisQha LuCiiajjaBelum ada peringkat
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (895)
- 5045Dokumen8 halaman5045SisQha LuCiiajjaBelum ada peringkat
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (400)
- Jurnal ImunologiDokumen7 halamanJurnal ImunologiSisQha LuCiiajjaBelum ada peringkat
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- Chrom MethodDokumen32 halamanChrom Methodmime84Belum ada peringkat
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Capillary Ion-Exchange Chromatography With Nanogram Sensitivity For The Analysis of Monoclonal AntibodiesDokumen1 halamanCapillary Ion-Exchange Chromatography With Nanogram Sensitivity For The Analysis of Monoclonal AntibodiesSisQha LuCiiajjaBelum ada peringkat
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- Novel Drug Delivery System Based On Docetaxel-Loaded Nanocapsules As A Therapeutic Strategy Against Breast Cancer CellsDokumen14 halamanNovel Drug Delivery System Based On Docetaxel-Loaded Nanocapsules As A Therapeutic Strategy Against Breast Cancer CellsSisQha LuCiiajjaBelum ada peringkat
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (588)
- Unimed Research 22447 66 Bab IIDokumen7 halamanUnimed Research 22447 66 Bab IISisQha LuCiiajjaBelum ada peringkat
- Validasi Metode Analisis Mikrobiologi s2.Dokumen59 halamanValidasi Metode Analisis Mikrobiologi s2.SisQha LuCiiajjaBelum ada peringkat
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- Jurnal Spo Baru 2Dokumen7 halamanJurnal Spo Baru 2SisQha LuCiiajjaBelum ada peringkat
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (74)
- Nanocochelates 2Dokumen7 halamanNanocochelates 2SisQha LuCiiajjaBelum ada peringkat
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
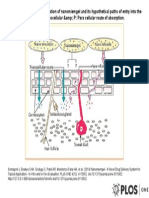
- Journal Pone 0115952 g001Dokumen1 halamanJournal Pone 0115952 g001SisQha LuCiiajja100% (1)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (266)
- Jchps 7 (3) 7 Kishore Babu 1 205-209Dokumen5 halamanJchps 7 (3) 7 Kishore Babu 1 205-209SisQha LuCiiajjaBelum ada peringkat
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- 1 s2.0 S0169409X14000696 MainDokumen14 halaman1 s2.0 S0169409X14000696 MainSisQha LuCiiajjaBelum ada peringkat
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- Validasi Metode Analisis Mikrobiologi s2.Dokumen59 halamanValidasi Metode Analisis Mikrobiologi s2.SisQha LuCiiajjaBelum ada peringkat
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2259)
- Bhatt Ganesh, Raturi Ankita & Kothiyal Preeti Shri Guru Ram Rai Institute of Technology & Sciences Dehradun, Uttarakhand, IndiaDokumen18 halamanBhatt Ganesh, Raturi Ankita & Kothiyal Preeti Shri Guru Ram Rai Institute of Technology & Sciences Dehradun, Uttarakhand, IndiaSisQha LuCiiajjaBelum ada peringkat
- ITS Paper 19992 PaperpdfpdfDokumen7 halamanITS Paper 19992 PaperpdfpdfSisQha LuCiiajjaBelum ada peringkat
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- Fentanyl inDokumen10 halamanFentanyl inSisQha LuCiiajjaBelum ada peringkat
- Validasi Metode Analisis Mikrobiologi s2.Dokumen59 halamanValidasi Metode Analisis Mikrobiologi s2.SisQha LuCiiajjaBelum ada peringkat
- Unimed Research 22447 66 Bab IIIDokumen8 halamanUnimed Research 22447 66 Bab IIISisQha LuCiiajjaBelum ada peringkat
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- Jurnal Susu Pasteurisasi BIOKIMDokumen6 halamanJurnal Susu Pasteurisasi BIOKIMAgung M OdonkBelum ada peringkat
- Unimed Research 22447 66 Bab IDokumen3 halamanUnimed Research 22447 66 Bab ISisQha LuCiiajjaBelum ada peringkat
- Validasi Metode Analisis Mikrobiologi s2.Dokumen59 halamanValidasi Metode Analisis Mikrobiologi s2.SisQha LuCiiajjaBelum ada peringkat
- Unimed Research 22447 66 Bab IDokumen3 halamanUnimed Research 22447 66 Bab ISisQha LuCiiajjaBelum ada peringkat
- 1 s2.0 S0169409X14000696 MainDokumen14 halaman1 s2.0 S0169409X14000696 MainSisQha LuCiiajjaBelum ada peringkat
- International Journal of Pharmacy and Pharmaceutical SciencesDokumen7 halamanInternational Journal of Pharmacy and Pharmaceutical SciencesSisQha LuCiiajjaBelum ada peringkat
- A Jps 20121072Dokumen13 halamanA Jps 20121072SisQha LuCiiajjaBelum ada peringkat
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (121)
- 1 s2.0 S0169409X14000696 MainDokumen14 halaman1 s2.0 S0169409X14000696 MainSisQha LuCiiajjaBelum ada peringkat
- GR 15 Juan Esteban Berrio Cordoba Guia 4 ENVIRONMENTAL PROBLEMSDokumen7 halamanGR 15 Juan Esteban Berrio Cordoba Guia 4 ENVIRONMENTAL PROBLEMSJuan Esteban Berrio CordobaBelum ada peringkat
- SurveyDokumen1 halamanSurveyJainne Ann BetchaidaBelum ada peringkat
- MSW TACSOP v.3.3Dokumen59 halamanMSW TACSOP v.3.3Mira BellaBelum ada peringkat
- Ejercicio 1 Curso BRBDokumen2 halamanEjercicio 1 Curso BRBAlex MolinaBelum ada peringkat
- 5 Natural Sore Throat RemediesDokumen5 halaman5 Natural Sore Throat Remedieslisa smithis100% (1)
- CAST Optical Smoke Detector: Part NoDokumen3 halamanCAST Optical Smoke Detector: Part NoAnis TantoushBelum ada peringkat
- Convecc Ao Forc Ada Externa: Vicente Luiz ScalonDokumen18 halamanConvecc Ao Forc Ada Externa: Vicente Luiz ScalonMaria VitóriaBelum ada peringkat
- Juri Ferrer - Ws - WeatherDokumen4 halamanJuri Ferrer - Ws - WeathersJIqsBelum ada peringkat
- AssaultDokumen12 halamanAssaultEd Mundo100% (3)
- Manual Servicio Soxte 2050Dokumen46 halamanManual Servicio Soxte 2050Quimica JordanlabBelum ada peringkat
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- Job Description - Director of AdvancementDokumen1 halamanJob Description - Director of AdvancementCanterburyCambridgeBelum ada peringkat
- 1erTareaMicroscopíasLópez Marmolejo Clere MishellDokumen4 halaman1erTareaMicroscopíasLópez Marmolejo Clere Mishellclere02marmolejoBelum ada peringkat
- Thyroid OphthalmopathyDokumen59 halamanThyroid OphthalmopathyLavanya MadabushiBelum ada peringkat
- Ferrography/oil Analysis: An Excellent Condition Monitoring TechniqueDokumen5 halamanFerrography/oil Analysis: An Excellent Condition Monitoring Techniquedaniel zorroBelum ada peringkat
- Final Workshop Report On Value Addition and AgroprocessingDokumen31 halamanFinal Workshop Report On Value Addition and AgroprocessingBett K. BernardBelum ada peringkat
- Pathological Anatomy IntroDokumen27 halamanPathological Anatomy IntroJoiya KhanBelum ada peringkat
- What Is Primary Health Care and Its Relation To Universal Health Care? As A Medical Student, What Impact Can I Create in Implementing PHC and UHC?Dokumen2 halamanWhat Is Primary Health Care and Its Relation To Universal Health Care? As A Medical Student, What Impact Can I Create in Implementing PHC and UHC?Aubrey Unique EvangelistaBelum ada peringkat
- Hookah Bar Business PlanDokumen34 halamanHookah Bar Business PlanAbdelkebir LabyadBelum ada peringkat
- Alix Pelletier Luginbühl - Works / PRINT VERSIONDokumen44 halamanAlix Pelletier Luginbühl - Works / PRINT VERSIONJonathan Philippe LEVYBelum ada peringkat
- Michigan Clinic 2008 NotesDokumen10 halamanMichigan Clinic 2008 NotesCoach Brown100% (3)
- PNOZ E1vp EN-23-27Dokumen5 halamanPNOZ E1vp EN-23-27Rachid MoussaouiBelum ada peringkat
- Telecommunications GroundingDokumen24 halamanTelecommunications GroundingMike FordealsBelum ada peringkat
- Zook Rupture Disc URADokumen2 halamanZook Rupture Disc URAmd_taheriBelum ada peringkat
- Vertical Transportation in BuildingsDokumen46 halamanVertical Transportation in BuildingsHIMA MiniBelum ada peringkat
- Outback MenuDokumen2 halamanOutback MenuzeeBelum ada peringkat
- DBM MonetizationDokumen2 halamanDBM MonetizationrsdiamzBelum ada peringkat
- Grades 4-10: Search Jobs in IndiaDokumen2 halamanGrades 4-10: Search Jobs in IndiaMD AKIL AHMEDBelum ada peringkat
- The Jewish Way in Death and MourningDokumen67 halamanThe Jewish Way in Death and Mourningizzy212100% (1)
- Rapid Review Hematology PDFDokumen155 halamanRapid Review Hematology PDFBaguma Michael100% (3)
- Complications of Diabetes MellitusDokumen46 halamanComplications of Diabetes MellitusAbhijith MenonBelum ada peringkat
- Sodium Bicarbonate: Nature's Unique First Aid RemedyDari EverandSodium Bicarbonate: Nature's Unique First Aid RemedyPenilaian: 5 dari 5 bintang5/5 (21)
- Process Plant Equipment: Operation, Control, and ReliabilityDari EverandProcess Plant Equipment: Operation, Control, and ReliabilityPenilaian: 5 dari 5 bintang5/5 (1)
- A New Approach to HAZOP of Complex Chemical ProcessesDari EverandA New Approach to HAZOP of Complex Chemical ProcessesBelum ada peringkat
- Guidelines for Chemical Process Quantitative Risk AnalysisDari EverandGuidelines for Chemical Process Quantitative Risk AnalysisPenilaian: 5 dari 5 bintang5/5 (1)
- Functional Safety from Scratch: A Practical Guide to Process Industry ApplicationsDari EverandFunctional Safety from Scratch: A Practical Guide to Process Industry ApplicationsBelum ada peringkat
- Physical and Chemical Equilibrium for Chemical EngineersDari EverandPhysical and Chemical Equilibrium for Chemical EngineersPenilaian: 5 dari 5 bintang5/5 (1)