Anda mungkin juga menyukai
- Direction Well Trajectory DesignDokumen10 halamanDirection Well Trajectory Designlaxmikant1983Belum ada peringkat
- Advanced Technology Solutions For Next Generation HPHT WellsDokumen15 halamanAdvanced Technology Solutions For Next Generation HPHT Wellsandrew sinagaBelum ada peringkat
- Modern Well Design: Second EditionDokumen5 halamanModern Well Design: Second Editionasdf123451812Belum ada peringkat
- Geol AbbreviationsDokumen212 halamanGeol AbbreviationsBikky KumarBelum ada peringkat
- Kappa CHL Book 5.40.02Dokumen366 halamanKappa CHL Book 5.40.02nghiaBelum ada peringkat
- Agbami 2014Dokumen1 halamanAgbami 2014Chiziterem Ndukwe-NwokeBelum ada peringkat
- EOR Methods GuideDokumen109 halamanEOR Methods GuideKarwan DilmanyBelum ada peringkat
- Sloughing Shale: by Anuj BhatiaDokumen9 halamanSloughing Shale: by Anuj BhatiaANUJ BHATIA100% (1)
- SPE-169245-MS Overcoming Ultradeepwater Cementing Challenges in The CaribbeanDokumen16 halamanSPE-169245-MS Overcoming Ultradeepwater Cementing Challenges in The CaribbeanAhmed Ali AlsubaihBelum ada peringkat
- Calculation of Formation Resistivity of Water From SP LogDokumen10 halamanCalculation of Formation Resistivity of Water From SP LogSushant Barge100% (1)
- Introduction To Petroleum GeologyDokumen10 halamanIntroduction To Petroleum GeologyDaniel Dadzie100% (1)
- GeologyDokumen34 halamanGeologyluanlubisBelum ada peringkat
- Oil Well DesigningDokumen15 halamanOil Well DesigningShashank SacamuriBelum ada peringkat
- Difficult Renal Puncture Geometrical StudyDokumen8 halamanDifficult Renal Puncture Geometrical StudyInternational Journal of Innovative Science and Research TechnologyBelum ada peringkat
- Coring and Core AnalysisDokumen63 halamanCoring and Core AnalysisSAMER SAMIRBelum ada peringkat
- WT AssignmentDokumen27 halamanWT Assignmentabdilrhman sulimanBelum ada peringkat
- Drillability of FormationsDokumen7 halamanDrillability of Formationsdrilling moneytreeBelum ada peringkat
- Overview of Drilling OperationDokumen106 halamanOverview of Drilling OperationTemitope BelloBelum ada peringkat
- Master's Thesis on Riserless Managed Pressure DrillingDokumen112 halamanMaster's Thesis on Riserless Managed Pressure DrillingRaul Blanco0% (1)
- 2 - Directional Well DrillingDokumen105 halaman2 - Directional Well DrillingAli AbdullahBelum ada peringkat
- Conference on IOR-EOR: Challenges, Process and Technologies in Indian Offshore FieldsDokumen31 halamanConference on IOR-EOR: Challenges, Process and Technologies in Indian Offshore Fieldsmch45616Belum ada peringkat
- PST0064 Practical HPHT Drilling and Well Engineering July2015Dokumen8 halamanPST0064 Practical HPHT Drilling and Well Engineering July2015JacobsBelum ada peringkat
- Coring ProgramsDokumen52 halamanCoring ProgramsNamwangala Rashid NatinduBelum ada peringkat
- L - 1 - INTRO - Well LoggingDokumen47 halamanL - 1 - INTRO - Well LoggingSaaeed Ali100% (1)
- 2015 ROP Enhancement in Shales Through Osmotic Processes SPE IADC 173138 MSDokumen19 halaman2015 ROP Enhancement in Shales Through Osmotic Processes SPE IADC 173138 MSSteven MarinoffBelum ada peringkat
- How Can We Produce When The Well Bore Is Cased and Cemented? OR How Will Production Arrive, If The Reservoir Zone Is Sealed or Cased Off?Dokumen21 halamanHow Can We Produce When The Well Bore Is Cased and Cemented? OR How Will Production Arrive, If The Reservoir Zone Is Sealed or Cased Off?crystalfaisal786Belum ada peringkat
- Well LoggingDokumen22 halamanWell LoggingSubham H Verma100% (1)
- WellFlo Group ProjectDokumen8 halamanWellFlo Group ProjectAngel NgoBelum ada peringkat
- Logging QuestionsDokumen11 halamanLogging QuestionsKarim HamdyBelum ada peringkat
- SPE-203872-MS - Multistage FracutringDokumen24 halamanSPE-203872-MS - Multistage Fracutringzach100% (1)
- Dipmeter MeasurementsDokumen18 halamanDipmeter Measurementsmahmoud_al_kafajiBelum ada peringkat
- Schlumberger LWD LoggerDokumen16 halamanSchlumberger LWD LoggeradityamduttaBelum ada peringkat
- ITPODokumen380 halamanITPOmadhuriBelum ada peringkat
- Kick ToleranceDokumen71 halamanKick ToleranceHafiz Othman100% (1)
- Master Thesis - Jose Maria MoratallaDokumen80 halamanMaster Thesis - Jose Maria MoratallaorlandoBelum ada peringkat
- Proppant Selection The Key To Successful Fracture StimulationDokumen10 halamanProppant Selection The Key To Successful Fracture Stimulationitalo ramirezBelum ada peringkat
- Modeling Optimizes Asset PerformanceDokumen5 halamanModeling Optimizes Asset Performanceapi-356706678100% (1)
- 1 - Introduction To Advanced Drilling EngineeringDokumen40 halaman1 - Introduction To Advanced Drilling EngineeringAli AbdullahBelum ada peringkat
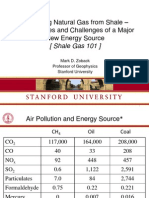
- MarkZoback ShaleGas101Dokumen93 halamanMarkZoback ShaleGas101Elan YogeswarenBelum ada peringkat
- Fit-For-Purpose Tubing-Conveyed Perforating Technique Increases Well Production by 250%Dokumen2 halamanFit-For-Purpose Tubing-Conveyed Perforating Technique Increases Well Production by 250%geokhan02Belum ada peringkat
- Directional Surveying Fundamentals and ToolsDokumen26 halamanDirectional Surveying Fundamentals and Toolsjesus alejandro lopez ovandoBelum ada peringkat
- PETE 625 Well Control: Lesson 1Dokumen37 halamanPETE 625 Well Control: Lesson 1Vandear GoalcantaraBelum ada peringkat
- Well StoryDokumen7 halamanWell Storyrslisbon100% (1)
- Question-1: Answer:: Convergent, Divergent and Transform Continental Margins Can Provide Oil & Gas?Dokumen6 halamanQuestion-1: Answer:: Convergent, Divergent and Transform Continental Margins Can Provide Oil & Gas?Shaker Ullah ChowdhuryBelum ada peringkat
- 1001 385v2-STIMPRO ISDokumen2 halaman1001 385v2-STIMPRO ISRodolfo PerezBelum ada peringkat
- ResChar-SEODokumen82 halamanResChar-SEOGerardo Zambrano PerdomoBelum ada peringkat
- Reservoir Engineering CPG 2020 3 - 2Dokumen120 halamanReservoir Engineering CPG 2020 3 - 2Chris ChijiokeBelum ada peringkat
- VICTORY VICTORY 1512032 Wells-Rosie Field 2638904 843650952Dokumen22 halamanVICTORY VICTORY 1512032 Wells-Rosie Field 2638904 843650952Agube VictoryBelum ada peringkat
- PETE403 FinalDokumen2 halamanPETE403 FinalZang NguyenBelum ada peringkat
- 1 1 PetroleumgeologyDokumen30 halaman1 1 PetroleumgeologyJEAN ALARCONBelum ada peringkat
- How to improve employee engagement and retentionDokumen299 halamanHow to improve employee engagement and retentionMehdi MehdiBelum ada peringkat
- SPE-124295-PP New Integrated Approach For Updating Pore-Pressure Predictions During DrillingDokumen11 halamanSPE-124295-PP New Integrated Approach For Updating Pore-Pressure Predictions During DrillingMarcelo Ayllón RiberaBelum ada peringkat
- DS Petrophysics - A4Dokumen8 halamanDS Petrophysics - A4hafideBelum ada peringkat
- Pre-Salt Presentation To KIVI Oct14 R2 PDFDokumen80 halamanPre-Salt Presentation To KIVI Oct14 R2 PDFmalakBelum ada peringkat
- Reservoir Evaluation Using Fall Off Testing and After Closure Analysis in the Khazzan FieldDokumen32 halamanReservoir Evaluation Using Fall Off Testing and After Closure Analysis in the Khazzan FieldNandani SudamaBelum ada peringkat
- AADE-17-NTCE-093 Common Misconceptions Regarding Lost Circulation TreatmentsDokumen5 halamanAADE-17-NTCE-093 Common Misconceptions Regarding Lost Circulation Treatmentsrobin0151Belum ada peringkat
- M SC Petroleum Engineer 2Dokumen3 halamanM SC Petroleum Engineer 2anouari2014Belum ada peringkat
- Final Round Toss-Up: Slug FlowDokumen4 halamanFinal Round Toss-Up: Slug FlowDeny Saputro ArifinBelum ada peringkat
- 2 Mechanisms Basin FormationDokumen24 halaman2 Mechanisms Basin FormationMuhamadKamilAzharBelum ada peringkat
- Hydrocarbon Fluid Inclusions in Petroliferous BasinsDari EverandHydrocarbon Fluid Inclusions in Petroliferous BasinsBelum ada peringkat
- Labmed31 0223Dokumen7 halamanLabmed31 0223GURUPRIYA ANANDBelum ada peringkat
- Causes, Symptoms and Treatments of Iron Deficiency AnemiaDokumen85 halamanCauses, Symptoms and Treatments of Iron Deficiency AnemiafrendirachmadBelum ada peringkat
- Thrombocytopenia2 PDFDokumen2 halamanThrombocytopenia2 PDFsafaggdafaBelum ada peringkat
- Hoff Et Al 1985 An Appraisal of Bone Marrow Biopsy in Assessment of Sick DogsDokumen9 halamanHoff Et Al 1985 An Appraisal of Bone Marrow Biopsy in Assessment of Sick DogsamnicolBelum ada peringkat
- White Blood Cell DisordersDokumen9 halamanWhite Blood Cell DisordersDocAxi Maximo Jr AxibalBelum ada peringkat
- Medicine MCQs For Medical Professionals - 3E (2013) (PDF)Dokumen933 halamanMedicine MCQs For Medical Professionals - 3E (2013) (PDF)Rohit Rajeev100% (14)
- Laboratory Tests for Diagnosing Anemia and OrganomegalyDokumen71 halamanLaboratory Tests for Diagnosing Anemia and OrganomegalyMedCoffee100% (1)
- Who Diagnostic Criteria MyelofibrosisDokumen1 halamanWho Diagnostic Criteria Myelofibrosispieterinpretoria391Belum ada peringkat
- MyelofibrosisDokumen22 halamanMyelofibrosisGd PadmawijayaBelum ada peringkat
- Myelofibrosis: Heri SutrisnoDokumen24 halamanMyelofibrosis: Heri SutrisnoDestrini Anjani LandaBelum ada peringkat
- Investigation and Management of Erythrocytosis: ReviewDokumen6 halamanInvestigation and Management of Erythrocytosis: ReviewCamilo CaceresBelum ada peringkat
- FS13 PolycythemiaVera FactSheetDokumen7 halamanFS13 PolycythemiaVera FactSheetMala 'emyu' UmarBelum ada peringkat
- Hematology EssentialsDokumen4 halamanHematology EssentialsAlfred ChowBelum ada peringkat
- WaloDokumen67 halamanWaloyepBelum ada peringkat
- Lab Studies of AnemiaDokumen13 halamanLab Studies of Anemiaeki1hidayatBelum ada peringkat
- White Blood Cell Disorders: Neoplastic Diseases of The BloodDokumen81 halamanWhite Blood Cell Disorders: Neoplastic Diseases of The BloodMiguel Cuevas Dolot100% (1)
- Hematopoietic SystemDokumen39 halamanHematopoietic SystemapplesncoreBelum ada peringkat
- Clinical Manifestations and Diagnosis of Primary MyelofibrosisDokumen7 halamanClinical Manifestations and Diagnosis of Primary MyelofibrosisVitor Hugo G CorreiaBelum ada peringkat
- Ask The Hematologist CompendiumDokumen51 halamanAsk The Hematologist Compendiumpieterinpretoria391Belum ada peringkat
- Clinical Approach To Isolated Splenomegaly: M M, P M, R KDokumen5 halamanClinical Approach To Isolated Splenomegaly: M M, P M, R KlilisBelum ada peringkat
- C+F-Essential Thrombocythemia in Dogs and Cats - Part IIDokumen9 halamanC+F-Essential Thrombocythemia in Dogs and Cats - Part IItaner_soysurenBelum ada peringkat
- ML7111 September quiz answers rapid referenceDokumen10 halamanML7111 September quiz answers rapid referenceCleo Salvador100% (2)
- PANCYTOPENIADokumen51 halamanPANCYTOPENIAResmyBelum ada peringkat
- 12345Dokumen73 halaman12345holly theressaBelum ada peringkat
- SysmexDokumen113 halamanSysmexARIF AHAMMED P100% (1)
- Oxford McqsDokumen15 halamanOxford McqsaliaaBelum ada peringkat
- New Med UpdatesDokumen88 halamanNew Med UpdatesMangesh ParadkarBelum ada peringkat
- ADA Guidelines 2016Dokumen14 halamanADA Guidelines 2016bbana1Belum ada peringkat
- Why Does My Patient Limphadenopathy or Splenomegaly?Dokumen14 halamanWhy Does My Patient Limphadenopathy or Splenomegaly?lacmftcBelum ada peringkat
- Aspek Laboratorium BCR Abl Kuantitatif & JAK2 V617FDokumen76 halamanAspek Laboratorium BCR Abl Kuantitatif & JAK2 V617FullifannuriBelum ada peringkat