Anda mungkin juga menyukai
- Pharmacoepidemiology, Pharmacoeconomics,PharmacovigilanceDari EverandPharmacoepidemiology, Pharmacoeconomics,PharmacovigilancePenilaian: 3 dari 5 bintang3/5 (1)
- Nonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsDari EverandNonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsWilliam J. BrockBelum ada peringkat
- Drug Discovery and Development HandoutDokumen5 halamanDrug Discovery and Development HandoutGermie PosionBelum ada peringkat
- A Review On Drug Approval in Regulated and Non-Regulated MarketsDokumen5 halamanA Review On Drug Approval in Regulated and Non-Regulated MarketsJohannes SchufiBelum ada peringkat
- 10 - Drug DevelopmentDokumen5 halaman10 - Drug DevelopmentLaura SaglietiBelum ada peringkat
- Resources For IND Applications: Back To TopDokumen4 halamanResources For IND Applications: Back To Topkavya nainitaBelum ada peringkat
- Investigational New DrugsDokumen16 halamanInvestigational New DrugsVirgil CendanaBelum ada peringkat
- New Drug Application: Presented By: Amey DeshpandeDokumen14 halamanNew Drug Application: Presented By: Amey Deshpandeamey_dpd100% (1)
- Medicine Price Surveys, Analyses and Comparisons: Evidence and Methodology GuidanceDari EverandMedicine Price Surveys, Analyses and Comparisons: Evidence and Methodology GuidanceSabine VoglerBelum ada peringkat
- Pharmacogenomics in Clinical TherapeuticsDari EverandPharmacogenomics in Clinical TherapeuticsLoralie J. LangmanBelum ada peringkat
- Current Good Manufacturing Practices (cGMP) for Pharmaceutical ProductsDari EverandCurrent Good Manufacturing Practices (cGMP) for Pharmaceutical ProductsBelum ada peringkat
- Chemical Sciences in Early Drug Discovery: Medicinal Chemistry 2.0Dari EverandChemical Sciences in Early Drug Discovery: Medicinal Chemistry 2.0Belum ada peringkat
- Food and Drug Regulation in an Era of Globalized MarketsDari EverandFood and Drug Regulation in an Era of Globalized MarketsSam F HalabiBelum ada peringkat
- Social Aspects of Drug Discovery, Development and CommercializationDari EverandSocial Aspects of Drug Discovery, Development and CommercializationBelum ada peringkat
- Ind Studies by Sejal KhumanDokumen34 halamanInd Studies by Sejal KhumanSejal khumamBelum ada peringkat
- Protocol Template - Early Phase.2Dokumen36 halamanProtocol Template - Early Phase.2Vasiliy KBelum ada peringkat
- Protocol Template 05feb2016 508Dokumen3 halamanProtocol Template 05feb2016 508Dwi Annisa AmaliaSariBelum ada peringkat
- FDA Guidance PMS and Clinical TrialsDokumen21 halamanFDA Guidance PMS and Clinical TrialsJonna SapiterBelum ada peringkat
- Pharmaco EpidemiologyDokumen3 halamanPharmaco EpidemiologyAnne RajeshBelum ada peringkat
- GMP ManualDokumen303 halamanGMP ManualShrinivas TamaskarBelum ada peringkat
- CTD CMC-Information-Human-Gene-Therapy-IND-Applications - Jan - 2020Dokumen56 halamanCTD CMC-Information-Human-Gene-Therapy-IND-Applications - Jan - 2020Shiraz KhanBelum ada peringkat
- An Introduction To Clinical Trials (Jonathan A. Cook)Dokumen277 halamanAn Introduction To Clinical Trials (Jonathan A. Cook)Nitin AnitaBelum ada peringkat
- Indonesia health sector pharmaceutical reformDokumen10 halamanIndonesia health sector pharmaceutical reformAna AsmaraBelum ada peringkat
- Clinical Guidelines Ecbs 2001Dokumen52 halamanClinical Guidelines Ecbs 2001Ajay KumarBelum ada peringkat
- Schedule YDokumen30 halamanSchedule Yapi-3810976100% (13)
- Abnormal Returns and Cash Flows in Pharmaceutical M&A PDFDokumen61 halamanAbnormal Returns and Cash Flows in Pharmaceutical M&A PDFJoachim_91Belum ada peringkat
- FDA Guidance For Industry S7a Safety Pharmacology Studies For Human Pharmaceuticals PDFDokumen14 halamanFDA Guidance For Industry S7a Safety Pharmacology Studies For Human Pharmaceuticals PDFbmartindoyle6396Belum ada peringkat
- Overview of Drug Discovery and Development ProcessesDokumen17 halamanOverview of Drug Discovery and Development ProcessesMelissa STanBelum ada peringkat
- EVALUATING CLINICAL Studies of Antimicrobials in The Division of Antiinfective Drug ProductsDokumen102 halamanEVALUATING CLINICAL Studies of Antimicrobials in The Division of Antiinfective Drug ProductsMichael wang100% (1)
- Clinical ResearchDokumen5 halamanClinical ResearchDeepti ShrivasBelum ada peringkat
- Ich GCPDokumen4 halamanIch GCPRaju GundaramBelum ada peringkat
- Maulik Shah Master Thesis DocumentDokumen107 halamanMaulik Shah Master Thesis DocumentchintanBelum ada peringkat
- Works For Sanofi AventisDokumen6 halamanWorks For Sanofi AventisMohit AroraBelum ada peringkat
- Dosage Form DesignDokumen19 halamanDosage Form DesignAnimikh RayBelum ada peringkat
- FDA Lab Presentation OverviewDokumen26 halamanFDA Lab Presentation OverviewAnupam ChoubeyBelum ada peringkat
- The Pharmaceutical Market Outlook To 2015 PDFDokumen131 halamanThe Pharmaceutical Market Outlook To 2015 PDFJosé Manuel Pais-ChanfrauBelum ada peringkat
- Portfolio, Program, and Project Management in the Pharmaceutical and Biotechnology IndustriesDari EverandPortfolio, Program, and Project Management in the Pharmaceutical and Biotechnology IndustriesPete HarpumBelum ada peringkat
- Careers in Focus: Pharmaceuticals and Biotechnology, Third EditionDari EverandCareers in Focus: Pharmaceuticals and Biotechnology, Third EditionBelum ada peringkat
- Senior Regulatory Affairs Specialist Pharmaceuticals in Philadelphia PA Resume Megan McKinneyDokumen2 halamanSenior Regulatory Affairs Specialist Pharmaceuticals in Philadelphia PA Resume Megan McKinneyMeganMcKinneyBelum ada peringkat
- 06 PSUR PBRER Thomas MunzDokumen35 halaman06 PSUR PBRER Thomas MunzMohabKamalBelum ada peringkat
- Topic 1 Pharma QUIZDokumen7 halamanTopic 1 Pharma QUIZJohn Daryl Sad-angBelum ada peringkat
- Types of Research TrialDokumen2 halamanTypes of Research TrialGems GlasgowBelum ada peringkat
- A Comparison of Pharmaceutical Promotional Tactics Between HK & ChinaDokumen10 halamanA Comparison of Pharmaceutical Promotional Tactics Between HK & ChinaAlfred LeungBelum ada peringkat
- Demand Diversity - COVID-19 ResearchDokumen39 halamanDemand Diversity - COVID-19 ResearchCOUCH Health50% (2)
- Biocontamination Control For Pharmaceuticals and HealthcareDokumen2 halamanBiocontamination Control For Pharmaceuticals and HealthcareTim Sandle100% (1)
- Pharmaceuticals Executive SummaryDokumen10 halamanPharmaceuticals Executive SummaryPradyot78Belum ada peringkat
- Novartis NVS Q4 2016 Ir PresentationDokumen84 halamanNovartis NVS Q4 2016 Ir PresentationmedtechyBelum ada peringkat
- CDSCODokumen14 halamanCDSCOAjay ModiBelum ada peringkat
- PHARMACEUTICAL PRODUCT DEVELOPMENT INC 10-K (Annual Reports) 2009-02-24Dokumen156 halamanPHARMACEUTICAL PRODUCT DEVELOPMENT INC 10-K (Annual Reports) 2009-02-24http://secwatch.com100% (2)
- Health Technology AssessmentDokumen8 halamanHealth Technology AssessmentJelcy MangulabnanBelum ada peringkat
- Understanding Clinical Trials: Developed by Sara Back, NP Bronx-Lebanon Hospital CenterDokumen41 halamanUnderstanding Clinical Trials: Developed by Sara Back, NP Bronx-Lebanon Hospital Centeranubha_naikBelum ada peringkat
- Regulatory Toxicology OverviewDokumen100 halamanRegulatory Toxicology OverviewHarsh KoshtiBelum ada peringkat
- Preclinical Trials PDFDokumen2 halamanPreclinical Trials PDFFiroz TPBelum ada peringkat
- IICRDokumen17 halamanIICRMukund SharmaBelum ada peringkat
- Drug Development: FDA's Definition of A New DrugDokumen11 halamanDrug Development: FDA's Definition of A New DrugSophie MendezBelum ada peringkat
- Clinical Trial DesignsDokumen18 halamanClinical Trial DesignsVenkatesh Gavini100% (1)
- Using Realworld Data For Outcomes Research and Comparative Effectiveness StudiesDokumen44 halamanUsing Realworld Data For Outcomes Research and Comparative Effectiveness StudiesTest Download100% (1)
- Expanding Access To Emergency Contraceptive Pills: Promoting Pharmacist/Prescriber Collaborative AgreementsDokumen57 halamanExpanding Access To Emergency Contraceptive Pills: Promoting Pharmacist/Prescriber Collaborative AgreementsRobert CantemprateBelum ada peringkat
- Solman SyllabusDokumen1 halamanSolman Syllabussapis18Belum ada peringkat
- Quality Center LogonDokumen1 halamanQuality Center Logonsapis18Belum ada peringkat
- Excel 2010 Undex 3Dokumen1 halamanExcel 2010 Undex 3sapis18Belum ada peringkat
- HPQC GuidelinesDokumen1 halamanHPQC Guidelinessapis18Belum ada peringkat
- SAP Solman InfrastructureDokumen1 halamanSAP Solman Infrastructuresapis18Belum ada peringkat
- Cap - MGMT in SapDokumen1 halamanCap - MGMT in Sapsapis18Belum ada peringkat
- SAP Solman Overview: Current Version of SAP Solution Manager and Release DateDokumen1 halamanSAP Solman Overview: Current Version of SAP Solution Manager and Release Datesapis18Belum ada peringkat
- Solman ProjectDokumen1 halamanSolman Projectsapis18Belum ada peringkat
- SAP Solman Work CenterDokumen1 halamanSAP Solman Work Centersapis18Belum ada peringkat
- PAS-X 2017 Training Program ModulesDokumen1 halamanPAS-X 2017 Training Program Modulessapis18Belum ada peringkat
- Capacity Planning Syllabus PDFDokumen1 halamanCapacity Planning Syllabus PDFsapis18Belum ada peringkat
- Capacity Planning Contents PDFDokumen1 halamanCapacity Planning Contents PDFsapis18Belum ada peringkat
- Capacity Planning ContentsDokumen1 halamanCapacity Planning Contentssapis18Belum ada peringkat
- Capcity Planning EbiteDokumen1 halamanCapcity Planning Ebitesapis18Belum ada peringkat
- Werum PAS X Training 2017 3Dokumen1 halamanWerum PAS X Training 2017 3sapis180% (1)
- Department of Communicat.Dokumen2 halamanDepartment of Communicat.sapis18Belum ada peringkat
- Werum PAS X Training 2017 2Dokumen1 halamanWerum PAS X Training 2017 2sapis180% (1)
- What Is Production PlanningDokumen1 halamanWhat Is Production Planningsapis18Belum ada peringkat
- 7 Deohri&RqwhqwvDokumen1 halaman7 Deohri&Rqwhqwvsapis18Belum ada peringkat
- Batch DeterminationDokumen1 halamanBatch Determinationsapis18Belum ada peringkat
- Sap S - Hana FinanceDokumen1 halamanSap S - Hana Financesapis18Belum ada peringkat
- Profitability Analysis in SAP S - HANA 1Dokumen1 halamanProfitability Analysis in SAP S - HANA 1sapis18Belum ada peringkat
- What Is The SOP ProcessDokumen1 halamanWhat Is The SOP Processsapis18Belum ada peringkat
- Activation and Prerequisites: Hana Features/ImportanceDokumen1 halamanActivation and Prerequisites: Hana Features/Importancesapis18Belum ada peringkat
- PP Processes and RolesSalesDokumen1 halamanPP Processes and RolesSalessapis18Belum ada peringkat
- Gatp Works Process - 1Dokumen1 halamanGatp Works Process - 1sapis18Belum ada peringkat
- Sap Legacy SystemDokumen1 halamanSap Legacy Systemsapis18Belum ada peringkat
- Customizing Your Quick Access ToolbarDokumen1 halamanCustomizing Your Quick Access Toolbarsapis18Belum ada peringkat
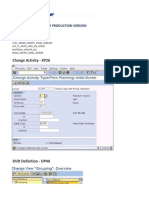
- Function Modules For Production VersionDokumen1 halamanFunction Modules For Production Versionsapis18Belum ada peringkat
- Md04 and Md05Dokumen1 halamanMd04 and Md05sapis18Belum ada peringkat
- White Paper: Where Is The SLP?Dokumen9 halamanWhite Paper: Where Is The SLP?thehellofoundBelum ada peringkat
- Chapter 1 Problem and Its ScopeDokumen9 halamanChapter 1 Problem and Its ScopeKhymberly Krystyn83% (12)
- Respondent Final Memo-NowDokumen27 halamanRespondent Final Memo-Nowimran alamBelum ada peringkat
- Medicolegal Aspects of Anaesthesia and Dilemmas To AnaesthetistDokumen58 halamanMedicolegal Aspects of Anaesthesia and Dilemmas To AnaesthetistVaibhav JainBelum ada peringkat
- Audit Observations and Recommendations on Gender and Development FundsDokumen43 halamanAudit Observations and Recommendations on Gender and Development FundsJonson PalmaresBelum ada peringkat
- REPORTING MEDICAL INCIDENTSDokumen2 halamanREPORTING MEDICAL INCIDENTSmale nurseBelum ada peringkat
- PP T On SNADokumen18 halamanPP T On SNADrPreeti Thakur ChouhanBelum ada peringkat
- Application for Disability Parking Placard and License PlateDokumen2 halamanApplication for Disability Parking Placard and License PlateKimberly AndrzejewskiBelum ada peringkat
- Final E-Office SopDokumen36 halamanFinal E-Office SopShadab AhmadBelum ada peringkat
- Laboratory OfficerDokumen8 halamanLaboratory OfficerTim DoustBelum ada peringkat
- Miller's Presentation To CommissionersDokumen15 halamanMiller's Presentation To CommissionersJennifer BowmanBelum ada peringkat
- EQ-5D-5L UserGuide 2015Dokumen28 halamanEQ-5D-5L UserGuide 2015Ioannis AmanatidisBelum ada peringkat
- CLEA 2015 (ASIA-INDIA) RespondentsDokumen36 halamanCLEA 2015 (ASIA-INDIA) RespondentsAmol MehtaBelum ada peringkat
- Over Booking: Hotel Overbooking Ways For A Hotel To Refuse You A RoomDokumen7 halamanOver Booking: Hotel Overbooking Ways For A Hotel To Refuse You A RoomoverbookBelum ada peringkat
- Gems Employment Contract ProposalDokumen7 halamanGems Employment Contract ProposalSatyendraYadavBelum ada peringkat
- Child Early and Forced MarriageDokumen148 halamanChild Early and Forced MarriageInter-Parliamentary Union100% (1)
- Mission Health-HCA Healthcare Inc. Asset Purchase Agreement, Aug. 30, 2018Dokumen147 halamanMission Health-HCA Healthcare Inc. Asset Purchase Agreement, Aug. 30, 2018Dillon DavisBelum ada peringkat
- Maxicare-Affiliated Providers - DOH-Certified Laboratories For COVID-19 Testing As of Sept 29, 2020Dokumen2 halamanMaxicare-Affiliated Providers - DOH-Certified Laboratories For COVID-19 Testing As of Sept 29, 2020Mj BoteroBelum ada peringkat
- Mfds Application Form DynamicDokumen2 halamanMfds Application Form DynamicZain AyubBelum ada peringkat
- Brandie Conrad counseling infoDokumen2 halamanBrandie Conrad counseling infoMarianLaloBelum ada peringkat
- Bibliography on Urban Waste ManagementDokumen45 halamanBibliography on Urban Waste ManagementRavikar ShyamBelum ada peringkat
- Canada Tobacco-Related LitigationDokumen29 halamanCanada Tobacco-Related LitigationIndonesia TobaccoBelum ada peringkat
- DFDokumen5 halamanDFDirajen PMBelum ada peringkat
- Teaching Billing and Coding To Medical Students: A Pilot StudyDokumen3 halamanTeaching Billing and Coding To Medical Students: A Pilot StudyafsdsaadsBelum ada peringkat
- Final Resume Package Douglas ShawDokumen3 halamanFinal Resume Package Douglas Shawapi-347691125Belum ada peringkat
- Moore v. Regents of The University of CalifoDokumen2 halamanMoore v. Regents of The University of Califodondz100% (1)
- Resident AgreementDokumen3 halamanResident Agreementbartoncreekal100% (2)
- Review of Related Literature on Solid Waste Management in the PhilippinesDokumen5 halamanReview of Related Literature on Solid Waste Management in the PhilippinesEureka Margreith Arro90% (10)
- Treasury Financial Manual 11-08Dokumen57 halamanTreasury Financial Manual 11-08topramenBelum ada peringkat
- Skipp Complaint To DPH Re: Dr. Sidney HorowitzDokumen58 halamanSkipp Complaint To DPH Re: Dr. Sidney HorowitzJournalistABC100% (3)