Anda mungkin juga menyukai
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- Coordination Chemistry Reviews: The Highest Oxidation States of The Transition Metal ElementsDokumen19 halamanCoordination Chemistry Reviews: The Highest Oxidation States of The Transition Metal ElementsMatthew JenkinsBelum ada peringkat
- Mercury Is A Transition Metal: The First Experimental Evidence For HGFDokumen5 halamanMercury Is A Transition Metal: The First Experimental Evidence For HGFMatthew JenkinsBelum ada peringkat
- IN001087Dokumen1 halamanIN001087Matthew JenkinsBelum ada peringkat
- Paper-10kV PCD PoznanSLR PDFDokumen6 halamanPaper-10kV PCD PoznanSLR PDFMatthew JenkinsBelum ada peringkat
- 10.1038@s41586 020 2171 6 PDFDokumen17 halaman10.1038@s41586 020 2171 6 PDFMatthew JenkinsBelum ada peringkat
- U:: ' '-: Li-', - :'J:?Ui G',AND-RF.C?RDSHEET:, I"'J, H' F."JDokumen702 halamanU:: ' '-: Li-', - :'J:?Ui G',AND-RF.C?RDSHEET:, I"'J, H' F."JC.D. StelzerBelum ada peringkat
- 10.1038@s41586 020 2171 6 PDFDokumen17 halaman10.1038@s41586 020 2171 6 PDFMatthew JenkinsBelum ada peringkat
- LabCorp COVID EUAsum3Dokumen12 halamanLabCorp COVID EUAsum3Matthew JenkinsBelum ada peringkat
- Against The Law: Countering Lawful Abuses of Digital SurveillanceDokumen16 halamanAgainst The Law: Countering Lawful Abuses of Digital SurveillanceSoftpediaBelum ada peringkat
- Fnbeh 12 00242 PDFDokumen16 halamanFnbeh 12 00242 PDFMatthew JenkinsBelum ada peringkat
- The FDTD Grid and The Yee Algorithm PDFDokumen41 halamanThe FDTD Grid and The Yee Algorithm PDFMatthew JenkinsBelum ada peringkat
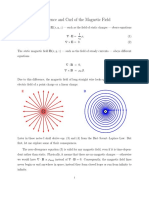
- Divergence and Curl of The Magnetic FieldDokumen6 halamanDivergence and Curl of The Magnetic FieldMatthew JenkinsBelum ada peringkat
- G. H. Faye A/-D E' H' Nickel "Dokumen10 halamanG. H. Faye A/-D E' H' Nickel "Matthew JenkinsBelum ada peringkat
- 1243 FullDokumen7 halaman1243 FullMatthew JenkinsBelum ada peringkat
- Seed Bank Contents ListDokumen1 halamanSeed Bank Contents ListMatthew JenkinsBelum ada peringkat
- 6th Central Pay Commission Salary CalculatorDokumen15 halaman6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- Susskind 2016Dokumen14 halamanSusskind 2016Matthew JenkinsBelum ada peringkat
- N Comms 10676Dokumen6 halamanN Comms 10676Matthew JenkinsBelum ada peringkat
- Hypotheses Regarding The Mechanisms of Ayahuasca in The Treatment of AddictionsDokumen10 halamanHypotheses Regarding The Mechanisms of Ayahuasca in The Treatment of AddictionsMatthew JenkinsBelum ada peringkat
- Borns Rule From Decoherence and EvarienceDokumen32 halamanBorns Rule From Decoherence and EvarienceWilliam HobbaBelum ada peringkat
- 6th Central Pay Commission Salary CalculatorDokumen15 halaman6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- Relative States and The Environment: Einselection, Envariance, Quantum Darwinism, and The Existential InterpretationDokumen27 halamanRelative States and The Environment: Einselection, Envariance, Quantum Darwinism, and The Existential InterpretationMatthew JenkinsBelum ada peringkat
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (895)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (400)
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (588)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (74)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (121)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- List of CAS Numbers by Chemical Compound - WikipediaDokumen84 halamanList of CAS Numbers by Chemical Compound - WikipediaFelipe Hdez LopezBelum ada peringkat
- Chapter 18 Metabolic PathwaysDokumen133 halamanChapter 18 Metabolic PathwaysM3hdi87Belum ada peringkat
- Biochemistry DKA NOTESDokumen278 halamanBiochemistry DKA NOTESTrisBelum ada peringkat
- Unit3 Stoichiometry QnsDokumen14 halamanUnit3 Stoichiometry QnsRanjan KathuriaBelum ada peringkat
- Evs 2 EcosystemDokumen27 halamanEvs 2 EcosystemSivakumar KBelum ada peringkat
- TIFR 2012 Solved PaperDokumen17 halamanTIFR 2012 Solved PaperMohit SoniBelum ada peringkat
- 11 Chemistry Solved Questions Chapter 8 PDFDokumen6 halaman11 Chemistry Solved Questions Chapter 8 PDFMohd UvaisBelum ada peringkat
- Bitsat Paper 03Dokumen21 halamanBitsat Paper 03Arnab SenBelum ada peringkat
- Study of Compounds - Ammonia A. State One Observation For The FollowingDokumen6 halamanStudy of Compounds - Ammonia A. State One Observation For The FollowingVishal SBelum ada peringkat
- 10+2 Chem P-Block ElementsDokumen44 halaman10+2 Chem P-Block ElementsArjun PasrichaBelum ada peringkat
- Odor Control: Wastewater Treatment/Odor Control Methods Odor Control SystemDokumen21 halamanOdor Control: Wastewater Treatment/Odor Control Methods Odor Control SystemRB MortxBelum ada peringkat
- Report 1Dokumen16 halamanReport 1Tuyết TrânBelum ada peringkat
- Inorganic Chemistry SynopsisDokumen24 halamanInorganic Chemistry SynopsisAvi KedarrBelum ada peringkat
- WINSEM2020-21 CHY1701 ELA VL2020210505041 Reference Material I 04-Mar-2021 7. Iron in Carbon Steel by PotentiometryDokumen6 halamanWINSEM2020-21 CHY1701 ELA VL2020210505041 Reference Material I 04-Mar-2021 7. Iron in Carbon Steel by PotentiometryAnurag karkiBelum ada peringkat
- AdeziuneDokumen13 halamanAdeziuneSintea Sorin-RobertinoBelum ada peringkat
- Corrosion EngineeringDokumen27 halamanCorrosion EngineeringMỹ Linh Lê100% (1)
- ChemistryDokumen10 halamanChemistrykahgua0% (1)
- Highly Porous Electrospun Polyvinylidene Fluoride (PVDF) - Based Carbon FiberDokumen9 halamanHighly Porous Electrospun Polyvinylidene Fluoride (PVDF) - Based Carbon FiberTeddy KimBelum ada peringkat
- Experiment-3: Galvanic SeriesDokumen4 halamanExperiment-3: Galvanic SeriesChayon MondalBelum ada peringkat
- CorrosionDokumen55 halamanCorrosionfreeuser3Belum ada peringkat
- 2023 o Level Chemistry SyllabusDokumen56 halaman2023 o Level Chemistry SyllabusFooxBelum ada peringkat
- Amination by ReductionDokumen46 halamanAmination by ReductionSidra LiaquatBelum ada peringkat
- Electrochemistry: (S) 2 2 (Aq) 2 (G)Dokumen30 halamanElectrochemistry: (S) 2 2 (Aq) 2 (G)Edon BediBelum ada peringkat
- Cyclic VoltammetryDokumen32 halamanCyclic VoltammetryAneeqa YounasBelum ada peringkat
- W VCE 2019 Chemistry Oxidation Numbers Worksheet 1 FINALDokumen4 halamanW VCE 2019 Chemistry Oxidation Numbers Worksheet 1 FINALhtyhongBelum ada peringkat
- Environmental Chemistry Notes: Compiled and Edited by Sir Layan 0774 372 589Dokumen14 halamanEnvironmental Chemistry Notes: Compiled and Edited by Sir Layan 0774 372 589maxwell Mutare100% (1)
- Baed-Chem2122-2212s Q2 Performance Task 3Dokumen5 halamanBaed-Chem2122-2212s Q2 Performance Task 3leyt kanaBelum ada peringkat
- Spare IcapDokumen18 halamanSpare Icapjonz afashBelum ada peringkat
- Cambridge International General Certificate of Secondary EducationDokumen16 halamanCambridge International General Certificate of Secondary EducationAbdulBasitBilalSheikhBelum ada peringkat
- BaseCatalyzedOxidation ReductionofAldehydesbytheCannizzaroReactionDokumen4 halamanBaseCatalyzedOxidation ReductionofAldehydesbytheCannizzaroReactionMike TranBelum ada peringkat