Anda mungkin juga menyukai
- Introduction To UltravioletVisible Molecular Absorption SpectrometryDokumen19 halamanIntroduction To UltravioletVisible Molecular Absorption SpectrometryMark Cliffton BadlonBelum ada peringkat
- Period (Year Started - Year Ended) Field University/ School Scholarship (If Applicable) RemarksDokumen3 halamanPeriod (Year Started - Year Ended) Field University/ School Scholarship (If Applicable) RemarksMark Cliffton BadlonBelum ada peringkat
- Introduction To Spectroscopic Methods: Instrumental AnalysisDokumen27 halamanIntroduction To Spectroscopic Methods: Instrumental AnalysisMark Cliffton BadlonBelum ada peringkat
- Gas Chromatography Not MineDokumen28 halamanGas Chromatography Not MineMark Cliffton BadlonBelum ada peringkat
- Introduction & Applications of Infrared SpectrometryDokumen20 halamanIntroduction & Applications of Infrared SpectrometryMark Cliffton BadlonBelum ada peringkat
- Introduction To Infrared SpectrosDokumen18 halamanIntroduction To Infrared SpectrosMark Cliffton BadlonBelum ada peringkat
- Gas Chromatography Not MineDokumen14 halamanGas Chromatography Not MineMark Cliffton BadlonBelum ada peringkat
- Electrical Components and Circuits Not MineDokumen21 halamanElectrical Components and Circuits Not MineMark Cliffton BadlonBelum ada peringkat
- FTIR SOP Not MineDokumen1 halamanFTIR SOP Not MineMark Cliffton BadlonBelum ada peringkat
- Electrolyte Effects Activity or Concentration Not MineDokumen14 halamanElectrolyte Effects Activity or Concentration Not MineMark Cliffton BadlonBelum ada peringkat
- Classification of Analytical Methods Not MineDokumen20 halamanClassification of Analytical Methods Not MineMark Cliffton BadlonBelum ada peringkat
- Determination of Optimum Flow Rate in Gas Chromatography Not MineDokumen2 halamanDetermination of Optimum Flow Rate in Gas Chromatography Not MineMark Cliffton BadlonBelum ada peringkat
- Chemicals and Apparatus Not MineDokumen12 halamanChemicals and Apparatus Not MineMark Cliffton BadlonBelum ada peringkat
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (400)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (588)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (266)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (74)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (121)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- PR KehumasanDokumen14 halamanPR KehumasanImamBelum ada peringkat
- WL-80 FTCDokumen5 halamanWL-80 FTCMr.Thawatchai hansuwanBelum ada peringkat
- MASONRYDokumen8 halamanMASONRYJowelyn MaderalBelum ada peringkat
- TCL LD24D50 - Chassis MS09A-LA - (TKLE2413D) - Manual de Servicio PDFDokumen41 halamanTCL LD24D50 - Chassis MS09A-LA - (TKLE2413D) - Manual de Servicio PDFFabian OrtuzarBelum ada peringkat
- Köppen Climate Classification - Wikipedia, The Free EncyclopediaDokumen15 halamanKöppen Climate Classification - Wikipedia, The Free EncyclopediaAndreea Tataru StanciBelum ada peringkat
- Theory GraphDokumen23 halamanTheory GraphArthur CarabioBelum ada peringkat
- Ron Kangas - IoanDokumen11 halamanRon Kangas - IoanBogdan SoptereanBelum ada peringkat
- Blue Prism Data Sheet - Provisioning A Blue Prism Database ServerDokumen5 halamanBlue Prism Data Sheet - Provisioning A Blue Prism Database Serverreddy_vemula_praveenBelum ada peringkat
- Mark Garside Resume May 2014Dokumen3 halamanMark Garside Resume May 2014api-199955558Belum ada peringkat
- The Indonesia National Clean Development Mechanism Strategy StudyDokumen223 halamanThe Indonesia National Clean Development Mechanism Strategy StudyGedeBudiSuprayogaBelum ada peringkat
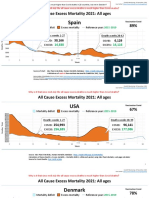
- Countries EXCESS DEATHS All Ages - 15nov2021Dokumen21 halamanCountries EXCESS DEATHS All Ages - 15nov2021robaksBelum ada peringkat
- Career Level Diagram - V5Dokumen1 halamanCareer Level Diagram - V5Shivani RaikwarBelum ada peringkat
- Chronic Kidney DiseaseDokumen15 halamanChronic Kidney Diseaseapi-270623039Belum ada peringkat
- Biological Beneficiation of Kaolin: A Review On Iron RemovalDokumen8 halamanBiological Beneficiation of Kaolin: A Review On Iron RemovalValentin GnoumouBelum ada peringkat
- Elements of ArtDokumen1 halamanElements of Artsamson8cindy8louBelum ada peringkat
- Review1 ScheduleDokumen3 halamanReview1 Schedulejayasuryam.ae18Belum ada peringkat
- ME Eng 8 Q1 0101 - SG - African History and LiteratureDokumen13 halamanME Eng 8 Q1 0101 - SG - African History and Literaturerosary bersanoBelum ada peringkat
- Article An Incident and Injury Free Culture Changing The Face of Project Operations Terra117 2Dokumen6 halamanArticle An Incident and Injury Free Culture Changing The Face of Project Operations Terra117 2nguyenthanhtuan_ecoBelum ada peringkat
- Existential ThreatsDokumen6 halamanExistential Threatslolab_4Belum ada peringkat
- II 2022 06 Baena-Rojas CanoDokumen11 halamanII 2022 06 Baena-Rojas CanoSebastian GaonaBelum ada peringkat
- KP Tevta Advertisement 16-09-2019Dokumen4 halamanKP Tevta Advertisement 16-09-2019Ishaq AminBelum ada peringkat
- Unit 16 - Monitoring, Review and Audit by Allan WatsonDokumen29 halamanUnit 16 - Monitoring, Review and Audit by Allan WatsonLuqman OsmanBelum ada peringkat
- 13 Adsorption of Congo Red A Basic Dye by ZnFe-CO3Dokumen10 halaman13 Adsorption of Congo Red A Basic Dye by ZnFe-CO3Jorellie PetalverBelum ada peringkat
- History of The Sikhs by Major Henry Cour PDFDokumen338 halamanHistory of The Sikhs by Major Henry Cour PDFDr. Kamalroop SinghBelum ada peringkat
- B. Geoinformatics PDFDokumen77 halamanB. Geoinformatics PDFmchakra720% (1)
- Mixed Up MonstersDokumen33 halamanMixed Up MonstersjaneBelum ada peringkat
- Stress-Strain Modelfor Grade275 Reinforcingsteel With Cyclic LoadingDokumen9 halamanStress-Strain Modelfor Grade275 Reinforcingsteel With Cyclic LoadingRory Cristian Cordero RojoBelum ada peringkat
- MGMT Audit Report WritingDokumen28 halamanMGMT Audit Report WritingAndrei IulianBelum ada peringkat
- Work ProblemsDokumen19 halamanWork ProblemsOfelia DavidBelum ada peringkat
- The JHipster Mini Book 2Dokumen129 halamanThe JHipster Mini Book 2tyulist100% (1)