Anda mungkin juga menyukai
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- VGEC: Teacher/Student Notes: Southern Blot ProtocolDokumen2 halamanVGEC: Teacher/Student Notes: Southern Blot ProtocolBi AnhBelum ada peringkat
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5795)
- Pset 2015Dokumen1 halamanPset 2015Bi AnhBelum ada peringkat
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (895)
- Biochimica Et Biophysica Acta: Jasvinder Singh Bhatti, Gurjit Kaur Bhatti, P. Hemachandra ReddyDokumen12 halamanBiochimica Et Biophysica Acta: Jasvinder Singh Bhatti, Gurjit Kaur Bhatti, P. Hemachandra ReddyBi AnhBelum ada peringkat
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- BS2009 Practicals 2008Dokumen4 halamanBS2009 Practicals 2008Bi AnhBelum ada peringkat
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (588)
- E00281 FullDokumen13 halamanE00281 FullBi AnhBelum ada peringkat
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (400)
- WWD 7.1 Discovery of A Second MessengerDokumen2 halamanWWD 7.1 Discovery of A Second MessengerBi AnhBelum ada peringkat
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- University of Toronto Ontario Biology Competition: 1995 Examination Answer KeyDokumen1 halamanUniversity of Toronto Ontario Biology Competition: 1995 Examination Answer KeyBi AnhBelum ada peringkat
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Theory and Measurement of Bacterial GrowthDokumen11 halamanTheory and Measurement of Bacterial GrowthBi Anh100% (1)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2259)
- Figure 7.9 Regulation of The Cell CycleDokumen1 halamanFigure 7.9 Regulation of The Cell CycleBi AnhBelum ada peringkat
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (74)
- WWD 7.1 Discovery of A Second MessengerDokumen2 halamanWWD 7.1 Discovery of A Second MessengerBi AnhBelum ada peringkat
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- History of Molecular BiologyHistory of Molecular Biology - Pdfhistory of Molecular Biology - Pdfvhistory of Molecular Biology PDFDokumen9 halamanHistory of Molecular BiologyHistory of Molecular Biology - Pdfhistory of Molecular Biology - Pdfvhistory of Molecular Biology PDFBi AnhBelum ada peringkat
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- BIOL207 Open GeneticsDokumen182 halamanBIOL207 Open GeneticsBi Anh100% (1)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- Exam 3 2008Dokumen9 halamanExam 3 2008Bi AnhBelum ada peringkat
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (266)
- UoN UnderGrad StudySkills 2011Dokumen184 halamanUoN UnderGrad StudySkills 2011Bi AnhBelum ada peringkat
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (345)
- How Cells Coordinate Growth and Division ReviewDokumen14 halamanHow Cells Coordinate Growth and Division ReviewBi AnhBelum ada peringkat
- Summer PlaceSummer Placements List Summer 2013ments List Summer 2013Dokumen21 halamanSummer PlaceSummer Placements List Summer 2013ments List Summer 2013Bi AnhBelum ada peringkat
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- Bio 306 General Physiology-2Dokumen143 halamanBio 306 General Physiology-2Bi AnhBelum ada peringkat
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- Section CDokumen1 halamanSection CBi AnhBelum ada peringkat
- BIOC7001 - Advanced DNA Techniques 2011 - FINALDokumen92 halamanBIOC7001 - Advanced DNA Techniques 2011 - FINALMao NanBelum ada peringkat
- Diffusion EssayDokumen2 halamanDiffusion Essayapi-242424506Belum ada peringkat
- ALVEOLAR BoneDokumen72 halamanALVEOLAR BoneArchana50% (2)
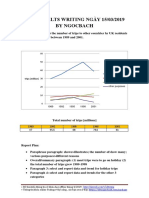
- Giai de Ielts Writing Ngay 150319 by NgocbachDokumen7 halamanGiai de Ielts Writing Ngay 150319 by NgocbachSedeaBelum ada peringkat
- John Toland - Pantheisticon PDFDokumen246 halamanJohn Toland - Pantheisticon PDFAlexander Elder100% (2)
- How To Grow Marijuana HydroponicallyDokumen33 halamanHow To Grow Marijuana Hydroponicallykim_peyoteBelum ada peringkat
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- CBSE Class 10 Life Processes MCQ Practice SolutionsDokumen14 halamanCBSE Class 10 Life Processes MCQ Practice SolutionsRAJESH BBelum ada peringkat
- Weaning Off Babies From A DiseaseDokumen272 halamanWeaning Off Babies From A DiseaseJun MagdaraogBelum ada peringkat
- CMT Lesson 2Dokumen14 halamanCMT Lesson 2Pam SyBelum ada peringkat
- Dna DissertationDokumen6 halamanDna DissertationCustomCollegePaperUK100% (1)
- Jump To Navigation Jump To Search: For Other Uses, SeeDokumen19 halamanJump To Navigation Jump To Search: For Other Uses, SeeDanny FentomBelum ada peringkat
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (121)
- SOP 3.04 DNA Extraction From BloodDokumen4 halamanSOP 3.04 DNA Extraction From BloodMeetali GuptaBelum ada peringkat
- AMICON Ultra Centrifuge DevicesDokumen9 halamanAMICON Ultra Centrifuge Devicesdjass1804Belum ada peringkat
- Spinal Cord and Spinal NervesDokumen9 halamanSpinal Cord and Spinal NervesJharaBelum ada peringkat
- Protimeter Timbermaster ManualDokumen5 halamanProtimeter Timbermaster ManualMakustiBelum ada peringkat
- Bio ElectrodesDokumen26 halamanBio ElectrodesSharok Nancy JoshuvaBelum ada peringkat
- TNSCST - OldDokumen15 halamanTNSCST - OldPG ChemistryBelum ada peringkat
- M. Beals, L. Gross, and S. Harrell (2000) - Diversity IndicesDokumen8 halamanM. Beals, L. Gross, and S. Harrell (2000) - Diversity Indiceslbayating100% (1)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- Restriction Enzymes: Restriction Enzymes Is An Enzyme That Cuts Double Stranded or SingleDokumen3 halamanRestriction Enzymes: Restriction Enzymes Is An Enzyme That Cuts Double Stranded or SingleArafat MiahBelum ada peringkat
- Reviews: Epidemiology, Definition and Treatment of Complicated Urinary Tract InfectionsDokumen15 halamanReviews: Epidemiology, Definition and Treatment of Complicated Urinary Tract InfectionsPutu AdiBelum ada peringkat
- D6595Dokumen6 halamanD6595moh_ichwanuddinBelum ada peringkat
- 1 Mobilising Ca2 to enhance fruit quality Preharvest application of Harpin αβ ProAct in citrus orchards in SpainDokumen7 halaman1 Mobilising Ca2 to enhance fruit quality Preharvest application of Harpin αβ ProAct in citrus orchards in SpainMurilo GrecoBelum ada peringkat
- Mark BartlettDokumen40 halamanMark BartlettbenedickBelum ada peringkat
- Solutions, Suspension and Colloidal SystemDokumen27 halamanSolutions, Suspension and Colloidal SystemmrvenkateshBelum ada peringkat
- 5 Enzymes: 0610 BiologyDokumen9 halaman5 Enzymes: 0610 BiologyAbdul HadiBelum ada peringkat
- Study of Amount of Casein in Different Samples of MilkDokumen9 halamanStudy of Amount of Casein in Different Samples of MilkPratyush Sharma80% (79)
- Polymers: A Comparative Review of Natural and Synthetic Biopolymer Composite ScaffoldsDokumen51 halamanPolymers: A Comparative Review of Natural and Synthetic Biopolymer Composite ScaffoldsAyesha ABelum ada peringkat
- Solving Material Balance Problems For Single Units Without ReactionsDokumen24 halamanSolving Material Balance Problems For Single Units Without ReactionsVanessa Arrieta HernándezBelum ada peringkat
- Embryonic Stem CellsDokumen3 halamanEmbryonic Stem CellsRajBelum ada peringkat
- When Somebody Loses Weight, Where Does The Fat Go ?Dokumen3 halamanWhen Somebody Loses Weight, Where Does The Fat Go ?Sílfide Xanat100% (1)
- Love Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Dari EverandLove Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Penilaian: 3 dari 5 bintang3/5 (1)
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedDari EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedPenilaian: 4.5 dari 5 bintang4.5/5 (82)
- ADHD is Awesome: A Guide to (Mostly) Thriving with ADHDDari EverandADHD is Awesome: A Guide to (Mostly) Thriving with ADHDPenilaian: 5 dari 5 bintang5/5 (3)