Anda mungkin juga menyukai
- Talking About Money: 1. DiscussDokumen4 halamanTalking About Money: 1. Discussjayweinx100% (1)
- How A Typical Five-Paragraph Essay Is Written SCDokumen4 halamanHow A Typical Five-Paragraph Essay Is Written SCjayweinxBelum ada peringkat
- It Must Be Halloween: 1. DiscussDokumen5 halamanIt Must Be Halloween: 1. DiscussjayweinxBelum ada peringkat
- F4 & 5 Modul Hebat BIODokumen142 halamanF4 & 5 Modul Hebat BIORouXuanChangBelum ada peringkat
- Novel: King Arthur: Theme: courage 勇敢Dokumen3 halamanNovel: King Arthur: Theme: courage 勇敢jayweinxBelum ada peringkat
- Writing The IntroductionDokumen3 halamanWriting The IntroductionjayweinxBelum ada peringkat
- The Scene at A Crowded Shopping MallDokumen4 halamanThe Scene at A Crowded Shopping MalljayweinxBelum ada peringkat
- My HeroDokumen7 halamanMy HerojayweinxBelum ada peringkat
- 体面Dokumen1 halaman体面jayweinxBelum ada peringkat
- January 1Dokumen49 halamanJanuary 1LAMCBelum ada peringkat
- f5 110319Dokumen3 halamanf5 110319jayweinxBelum ada peringkat
- CW Compilation (2017 Trials)Dokumen4 halamanCW Compilation (2017 Trials)FBBelum ada peringkat
- Report On Co-CurricularDokumen3 halamanReport On Co-CurricularjayweinxBelum ada peringkat
- Pep. Set 4 PT3 2016 PDFDokumen2 halamanPep. Set 4 PT3 2016 PDFjayweinx100% (1)
- Ujian Sumatif 1 Ting 1 2017Dokumen8 halamanUjian Sumatif 1 Ting 1 2017jayweinxBelum ada peringkat
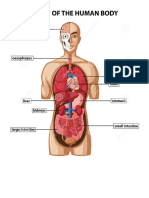
- Anatomy of Human BodyDokumen1 halamanAnatomy of Human BodyjayweinxBelum ada peringkat
- Mid Year f2 2016Dokumen14 halamanMid Year f2 2016jayweinxBelum ada peringkat
- Anatomy of Human BodyDokumen1 halamanAnatomy of Human BodyjayweinxBelum ada peringkat
- Directed Writing Report About The School CanteenDokumen4 halamanDirected Writing Report About The School Canteenjayweinx100% (2)
- E.) 5 Cba Heart AttackDokumen5 halamanE.) 5 Cba Heart AttackjayweinxBelum ada peringkat
- Line EeeeDokumen1 halamanLine EeeejayweinxBelum ada peringkat
- Ibmyp2 10Dokumen11 halamanIbmyp2 10jayweinxBelum ada peringkat
- Algebraic Expressions ExerciseDokumen18 halamanAlgebraic Expressions ExerciseLi Ann ChungBelum ada peringkat
- Modul 2 PDF July 13 2009 9 39 PM 181kDokumen10 halamanModul 2 PDF July 13 2009 9 39 PM 181kArun ManiamBelum ada peringkat
- Mathematics Notes Form 2: Ms. G. BonniciDokumen52 halamanMathematics Notes Form 2: Ms. G. BonnicijayweinxBelum ada peringkat
- Tips 2017 SPM EngDokumen13 halamanTips 2017 SPM EngjayweinxBelum ada peringkat
- English Test Written Test GrammarDokumen10 halamanEnglish Test Written Test Grammartarubi53100% (3)
- Chanyeol EXO Punch - Stay With MeDokumen3 halamanChanyeol EXO Punch - Stay With MejayweinxBelum ada peringkat
- f3 Bi Conjuction, Application LetterDokumen7 halamanf3 Bi Conjuction, Application LetterjayweinxBelum ada peringkat
- C1 Maths F1 Topical Test 1Dokumen3 halamanC1 Maths F1 Topical Test 1jayweinxBelum ada peringkat
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (400)
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (588)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (266)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (121)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- R265 FullDokumen13 halamanR265 FullMuch MahmudiBelum ada peringkat
- Gat B SyllabusDokumen3 halamanGat B SyllabusDIKCHHA AGRAWALBelum ada peringkat
- Biology of SNU Cell LinesDokumen19 halamanBiology of SNU Cell Lineshari_iyer16Belum ada peringkat
- 2nd - Part 3 - Lipid Structure and MetabolismDokumen56 halaman2nd - Part 3 - Lipid Structure and MetabolismAkbarWirawanBelum ada peringkat
- Nutrient Categories and FunctionsDokumen7 halamanNutrient Categories and Functionswiwin apri hartinaBelum ada peringkat
- Mol Bio Worksheet 1Dokumen3 halamanMol Bio Worksheet 1Wendell TulayBelum ada peringkat
- Rootcauseanalysis Invalid IC PCRDokumen6 halamanRootcauseanalysis Invalid IC PCRrpl5polbanBelum ada peringkat
- This Study Resource Was: DNA Base Pairing WorksheetDokumen5 halamanThis Study Resource Was: DNA Base Pairing Worksheetphil tolentinoBelum ada peringkat
- Chapter 3 Active Reading GuideDokumen10 halamanChapter 3 Active Reading GuideAnelaBelum ada peringkat
- DR Linda Molecular BiosDokumen73 halamanDR Linda Molecular BiosValency BathoBelum ada peringkat
- Websites Relating To Genetic EngineeringDokumen5 halamanWebsites Relating To Genetic Engineeringayie65Belum ada peringkat
- MAN0014394 - HighResMeltExperiment - GSG - Optimal HRMDokumen88 halamanMAN0014394 - HighResMeltExperiment - GSG - Optimal HRMphuoc dinhBelum ada peringkat
- The Organic Codes An Introduction To Semantic BiolDokumen3 halamanThe Organic Codes An Introduction To Semantic BiolIsmailBelum ada peringkat
- Calculation Examples Feeding Values For RuminantsDokumen8 halamanCalculation Examples Feeding Values For RuminantsВолодимир Сидоренко100% (1)
- NAAS Rating of JournalsDokumen44 halamanNAAS Rating of JournalssakshiBelum ada peringkat
- PhysioEx Exercise 8 Activity 2 - Balamad, Maria Karla M.Dokumen3 halamanPhysioEx Exercise 8 Activity 2 - Balamad, Maria Karla M.Maria Karla BalamadBelum ada peringkat
- Pulse-Chase Labeling of Protein Antigens With (S) MethionineDokumen7 halamanPulse-Chase Labeling of Protein Antigens With (S) MethionineSatt Michael Sobiono SantosBelum ada peringkat
- Bacterialand Archael InclusionsDokumen15 halamanBacterialand Archael InclusionsXimena AlcántaraBelum ada peringkat
- The SH2 Domain and Kinase Activity of JAK2 Target JAK2 To Centrosome and Regulate Cell Growth and Centrosome AmplificationDokumen22 halamanThe SH2 Domain and Kinase Activity of JAK2 Target JAK2 To Centrosome and Regulate Cell Growth and Centrosome AmplificationkshariqmBelum ada peringkat
- A Seminar Presentation On Mechanism of Drug ActionDokumen40 halamanA Seminar Presentation On Mechanism of Drug Actionbellatrix aliaBelum ada peringkat
- Name Pi R Group: Characteristic Three LetterDokumen1 halamanName Pi R Group: Characteristic Three LetterAloysius QuitaligBelum ada peringkat
- Bacteriology Lecture 2Dokumen63 halamanBacteriology Lecture 2Michelle Dy100% (2)
- Ion ChannelsDokumen22 halamanIon ChannelsGanesh V GaonkarBelum ada peringkat
- C2-Organisation and Maintenance of The OrganismDokumen39 halamanC2-Organisation and Maintenance of The OrganismyourmoBelum ada peringkat
- Biochemistry 7th Edition Berg Test BankDokumen35 halamanBiochemistry 7th Edition Berg Test Bankagleamamusable.pwclcq100% (21)
- Advanced Nutrition and Human Metabolism 7th Edition Gropper Solutions ManualDokumen19 halamanAdvanced Nutrition and Human Metabolism 7th Edition Gropper Solutions ManualMariaDaviesqrbg100% (39)
- Chapter 2 LipidsDokumen109 halamanChapter 2 LipidsTiffany Shane VallenteBelum ada peringkat
- Solution of Biology SSC-I (3rd Set)Dokumen13 halamanSolution of Biology SSC-I (3rd Set)Zunaira SafdarBelum ada peringkat
- The Amyloid Beta Peptide A Chemist S Perspective. Role in Alzheimer's and FibrillizationDokumen46 halamanThe Amyloid Beta Peptide A Chemist S Perspective. Role in Alzheimer's and FibrillizationAdrianEBelum ada peringkat
- Designing Insulin For Diabetes Therapy by Protein EngineeringDokumen7 halamanDesigning Insulin For Diabetes Therapy by Protein EngineeringJemiBelum ada peringkat