Anda mungkin juga menyukai
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (587)
- Aortic CoarctationDokumen9 halamanAortic CoarctationAustine OsaweBelum ada peringkat
- Hip FractureDokumen672 halamanHip FractureAustine OsaweBelum ada peringkat
- Sample Obs ReportDokumen1 halamanSample Obs ReportAustine OsaweBelum ada peringkat
- Coarctation of The Aorta: P. Syamasundar Rao, MDDokumen10 halamanCoarctation of The Aorta: P. Syamasundar Rao, MDAustine OsaweBelum ada peringkat
- Head InjuryDokumen61 halamanHead InjuryRicky Cornelius TariganBelum ada peringkat
- Dubin JDokumen2 halamanDubin JAustine OsaweBelum ada peringkat
- Reader in Orthopaedics, University of Glasgow: Roland BarnesDokumen11 halamanReader in Orthopaedics, University of Glasgow: Roland BarnesAustine OsaweBelum ada peringkat
- Aneurysmal Bone Cyst of Talus: A Rare CaseDokumen4 halamanAneurysmal Bone Cyst of Talus: A Rare CaseAustine OsaweBelum ada peringkat
- Aneurysmal Bone CystDokumen5 halamanAneurysmal Bone CystAustine OsaweBelum ada peringkat
- Dubin Johnson Synd.Dokumen4 halamanDubin Johnson Synd.Austine OsaweBelum ada peringkat
- Dubin Johnson Synd.Dokumen4 halamanDubin Johnson Synd.Austine OsaweBelum ada peringkat
- Dental Plaque As A Biofilm The Significance of PH in Health and Caries P MarshDokumen9 halamanDental Plaque As A Biofilm The Significance of PH in Health and Caries P MarshMuchlis Fauzi EBelum ada peringkat
- Aneurysmal Bone CystDokumen3 halamanAneurysmal Bone CystAustine OsaweBelum ada peringkat
- Severe jaundice case report highlights rare combination of Dubin-Johnson syndrome and hereditary spherocytosisDokumen4 halamanSevere jaundice case report highlights rare combination of Dubin-Johnson syndrome and hereditary spherocytosisAustine OsaweBelum ada peringkat
- Management of Proximal Humeral FracturesDokumen16 halamanManagement of Proximal Humeral FracturesAustine OsaweBelum ada peringkat
- Humeral Shaft Fracture GuideDokumen9 halamanHumeral Shaft Fracture GuideAustine Osawe100% (1)
- Burrows - OM Guideline MichiganDokumen12 halamanBurrows - OM Guideline MichiganLeo KolongBelum ada peringkat
- Hip FractureDokumen672 halamanHip FractureAustine OsaweBelum ada peringkat
- Hiperbilirubinemia by Ucsf HospitalDokumen3 halamanHiperbilirubinemia by Ucsf HospitalGalenica HoneyBelum ada peringkat
- Scleroderma PDFDokumen9 halamanScleroderma PDFAustine OsaweBelum ada peringkat
- Prevention of Rickets and Vitamin D Deficiency in Infants, Children, and AdolescentsDokumen13 halamanPrevention of Rickets and Vitamin D Deficiency in Infants, Children, and AdolescentsAustine OsaweBelum ada peringkat
- Dental PlaqueDokumen33 halamanDental PlaqueIbrahim AbdelHadiBelum ada peringkat
- BPJ PDFDokumen5 halamanBPJ PDFriyanBelum ada peringkat
- JaundiceDokumen12 halamanJaundicePatrick RamosBelum ada peringkat
- HistoplasmosisDokumen19 halamanHistoplasmosisapi-26188232100% (1)
- Coccidioidomycosis PDFDokumen7 halamanCoccidioidomycosis PDFAustine OsaweBelum ada peringkat
- Histoplasmosis: Fungal Disease Series #3Dokumen2 halamanHistoplasmosis: Fungal Disease Series #3Austine OsaweBelum ada peringkat
- Overdose and Poisoning in Adults 2006Dokumen7 halamanOverdose and Poisoning in Adults 2006Austine OsaweBelum ada peringkat
- His To Plasm OsisDokumen2 halamanHis To Plasm OsisAustine OsaweBelum ada peringkat
- Drug OverdoseDokumen2 halamanDrug OverdoseAustine OsaweBelum ada peringkat
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (894)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (399)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2219)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (265)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (119)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- How Psychology Has Changed Over TimeDokumen2 halamanHow Psychology Has Changed Over TimeMaedot HaddisBelum ada peringkat
- FSRH Ukmec Summary September 2019Dokumen11 halamanFSRH Ukmec Summary September 2019Kiran JayaprakashBelum ada peringkat
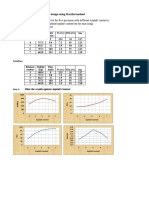
- Marshal HMA Mixture Design ExampleDokumen2 halamanMarshal HMA Mixture Design ExampleTewodros TadesseBelum ada peringkat
- ServiceDokumen47 halamanServiceMarko KoširBelum ada peringkat
- Bharhut Stupa Toraa Architectural SplenDokumen65 halamanBharhut Stupa Toraa Architectural Splenအသွ်င္ ေကသရBelum ada peringkat
- Evolution of Bluetooth PDFDokumen2 halamanEvolution of Bluetooth PDFJuzerBelum ada peringkat
- 1.2 - Venn Diagram and Complement of A SetDokumen6 halaman1.2 - Venn Diagram and Complement of A SetKaden YeoBelum ada peringkat
- Pub - Essentials of Nuclear Medicine Imaging 5th Edition PDFDokumen584 halamanPub - Essentials of Nuclear Medicine Imaging 5th Edition PDFNick Lariccia100% (1)
- Non Circumvention Non Disclosure Agreement (TERENCE) SGDokumen7 halamanNon Circumvention Non Disclosure Agreement (TERENCE) SGLin ChrisBelum ada peringkat
- DLP in Health 4Dokumen15 halamanDLP in Health 4Nina Claire Bustamante100% (1)
- PLC Networking with Profibus and TCP/IP for Industrial ControlDokumen12 halamanPLC Networking with Profibus and TCP/IP for Industrial Controltolasa lamessaBelum ada peringkat
- Good Ethics Is Good BusinessDokumen9 halamanGood Ethics Is Good BusinesssumeetpatnaikBelum ada peringkat
- Rubber Chemical Resistance Chart V001MAR17Dokumen27 halamanRubber Chemical Resistance Chart V001MAR17Deepak patilBelum ada peringkat
- TheEconomist 2023 04 01Dokumen297 halamanTheEconomist 2023 04 01Sh FBelum ada peringkat
- ASMOPS 2016 - International Invitation PHILIPPINEDokumen4 halamanASMOPS 2016 - International Invitation PHILIPPINEMl Phil0% (3)
- Complete Guide To Sports Training PDFDokumen105 halamanComplete Guide To Sports Training PDFShahana ShahBelum ada peringkat
- 4 Wheel ThunderDokumen9 halaman4 Wheel ThunderOlga Lucia Zapata SavaresseBelum ada peringkat
- Pom Final On Rice MillDokumen21 halamanPom Final On Rice MillKashif AliBelum ada peringkat
- KPMG Inpection ReportDokumen11 halamanKPMG Inpection ReportMacharia NgunjiriBelum ada peringkat
- Service and Maintenance Manual: Models 600A 600AJDokumen342 halamanService and Maintenance Manual: Models 600A 600AJHari Hara SuthanBelum ada peringkat
- Personalised MedicineDokumen25 halamanPersonalised MedicineRevanti MukherjeeBelum ada peringkat
- Google Dorks For PentestingDokumen11 halamanGoogle Dorks For PentestingClara Elizabeth Ochoa VicenteBelum ada peringkat
- Cold Rolled Steel Sections - Specification: Kenya StandardDokumen21 halamanCold Rolled Steel Sections - Specification: Kenya StandardPEng. Tech. Alvince KoreroBelum ada peringkat
- Rishte ki baat SMS messages collectionDokumen108 halamanRishte ki baat SMS messages collectionTushar AggarwalBelum ada peringkat
- Neuropsychological Deficits in Disordered Screen Use Behaviours - A Systematic Review and Meta-AnalysisDokumen32 halamanNeuropsychological Deficits in Disordered Screen Use Behaviours - A Systematic Review and Meta-AnalysisBang Pedro HattrickmerchBelum ada peringkat
- AFNOR IPTDS BrochureDokumen1 halamanAFNOR IPTDS Brochurebdiaconu20048672Belum ada peringkat
- Petty Cash Vouchers:: Accountability Accounted ForDokumen3 halamanPetty Cash Vouchers:: Accountability Accounted ForCrizhae OconBelum ada peringkat
- Radio Frequency Transmitter Type 1: System OperationDokumen2 halamanRadio Frequency Transmitter Type 1: System OperationAnonymous qjoKrp0oBelum ada peringkat
- FranklinDokumen4 halamanFranklinapi-291282463Belum ada peringkat
- Olympics Notes by Yousuf Jalal - PDF Version 1Dokumen13 halamanOlympics Notes by Yousuf Jalal - PDF Version 1saad jahangirBelum ada peringkat