Anda mungkin juga menyukai
- Tumor Necrosis Factor Alpha Influences The Inflammatory Response After Coronary SurgeryDokumen6 halamanTumor Necrosis Factor Alpha Influences The Inflammatory Response After Coronary Surgeryfluid_man_brazilBelum ada peringkat
- Long-Term Neurocognitive Function After Mechanical Aortic Valve ReplacementDokumen5 halamanLong-Term Neurocognitive Function After Mechanical Aortic Valve Replacementfluid_man_brazilBelum ada peringkat
- Attracting Outstanding Students (Premedical and Medical) To A Career in Cardiothoracic SurgeryDokumen3 halamanAttracting Outstanding Students (Premedical and Medical) To A Career in Cardiothoracic Surgeryfluid_man_brazilBelum ada peringkat
- Benefit of Partial Right-Bilateral Internal Thoracic Artery Harvesting in Patients at Risk of Sternal Wound ComplicationsDokumen5 halamanBenefit of Partial Right-Bilateral Internal Thoracic Artery Harvesting in Patients at Risk of Sternal Wound Complicationsfluid_man_brazilBelum ada peringkat
- (Radial String Sign) PDFDokumen8 halaman(Radial String Sign) PDFfluid_man_brazilBelum ada peringkat
- Wrapping of The Left Internal Thoracic Artery With An Expanded Polytetrafluoroethylene MembraneDokumen3 halamanWrapping of The Left Internal Thoracic Artery With An Expanded Polytetrafluoroethylene Membranefluid_man_brazilBelum ada peringkat
- Aprotinin Shows Both Hemostatic and Antithrombotic Effects During Off-Pump Coronary Artery Bypass GraftingDokumen8 halamanAprotinin Shows Both Hemostatic and Antithrombotic Effects During Off-Pump Coronary Artery Bypass Graftingfluid_man_brazilBelum ada peringkat
- Comparative Effects of Tolazoline and Nitroprusside On Human Isolated Radial ArteryDokumen7 halamanComparative Effects of Tolazoline and Nitroprusside On Human Isolated Radial Arteryfluid_man_brazilBelum ada peringkat
- Effects of Papaverine and Glycerilnitrate-Verapamil Solution As Topical and Intraluminal Vasodilators For Internal Thoracic ArteryDokumen5 halamanEffects of Papaverine and Glycerilnitrate-Verapamil Solution As Topical and Intraluminal Vasodilators For Internal Thoracic Arteryfluid_man_brazilBelum ada peringkat
- Personal Communication - A Piece of Glass in The HeartDokumen2 halamanPersonal Communication - A Piece of Glass in The Heartfluid_man_brazilBelum ada peringkat
- Off-Pump Myocardial Revascularization: Critical Analysis of 23 Years' Experience in 3,866 PatientsDokumen5 halamanOff-Pump Myocardial Revascularization: Critical Analysis of 23 Years' Experience in 3,866 Patientsfluid_man_brazilBelum ada peringkat
- The Society of Thoracic Surgeons/American Association For Thoracic Surgery Off-Pump Training ProgramDokumen3 halamanThe Society of Thoracic Surgeons/American Association For Thoracic Surgery Off-Pump Training Programfluid_man_brazilBelum ada peringkat
- Splanchnic Organ Injury During Coronary Surgery With or Without Cardiopulmonary Bypass: A Randomized, Controlled TrialDokumen7 halamanSplanchnic Organ Injury During Coronary Surgery With or Without Cardiopulmonary Bypass: A Randomized, Controlled Trialfluid_man_brazilBelum ada peringkat
- Myocardial Thievery: The Coronary-Subclavian Steal SyndromeDokumen7 halamanMyocardial Thievery: The Coronary-Subclavian Steal Syndromefluid_man_brazilBelum ada peringkat
- Physicians Assistants in Cardiothoracic Surgery: A 30-Year Experience in A University CenterDokumen6 halamanPhysicians Assistants in Cardiothoracic Surgery: A 30-Year Experience in A University Centerfluid_man_brazilBelum ada peringkat
- Coapsys Mitral Annuloplasty For Chronic Functional Ischemic Mitral Regurgitation: 1-Year ResultsDokumen5 halamanCoapsys Mitral Annuloplasty For Chronic Functional Ischemic Mitral Regurgitation: 1-Year Resultsfluid_man_brazilBelum ada peringkat
- Three-Dimensional Demonstration of The Collateral Circulation To The Artery of Adamkiewicz With 16-Row Multislice Computed TomographyDokumen1 halamanThree-Dimensional Demonstration of The Collateral Circulation To The Artery of Adamkiewicz With 16-Row Multislice Computed Tomographyfluid_man_brazilBelum ada peringkat
- Intraoperative and Postoperative Evaluation of Cavitation in Mechanical Heart Valve PatientsDokumen8 halamanIntraoperative and Postoperative Evaluation of Cavitation in Mechanical Heart Valve Patientsfluid_man_brazilBelum ada peringkat
- Invited Commentary - New Technology - New Perfusion System For Opcabg (Koshida Et Al)Dokumen2 halamanInvited Commentary - New Technology - New Perfusion System For Opcabg (Koshida Et Al)fluid_man_brazilBelum ada peringkat
- Coronary Artery Revascularization After Chest Wall Reconstruction With Rectus Abdominis Myocutaneous FlapDokumen4 halamanCoronary Artery Revascularization After Chest Wall Reconstruction With Rectus Abdominis Myocutaneous Flapfluid_man_brazilBelum ada peringkat
- How To Do It - Alternative Bi-Pectoral Muscle Flaps For Postoperative Sternotomy MediastinitisDokumen2 halamanHow To Do It - Alternative Bi-Pectoral Muscle Flaps For Postoperative Sternotomy Mediastinitisfluid_man_brazilBelum ada peringkat
- Early Postoperative Anticoagulation After Mechanical Valve Replacement: A Systematic ReviewDokumen12 halamanEarly Postoperative Anticoagulation After Mechanical Valve Replacement: A Systematic Reviewfluid_man_brazilBelum ada peringkat
- Images - Multiple Penetrating Aortic UlcersDokumen1 halamanImages - Multiple Penetrating Aortic Ulcersfluid_man_brazilBelum ada peringkat
- Portable Coronary Active Perfusion System For Off-Pump Coronary Artery Bypass GraftingDokumen5 halamanPortable Coronary Active Perfusion System For Off-Pump Coronary Artery Bypass Graftingfluid_man_brazilBelum ada peringkat
- Effect of Aprotinin and Recombinant Variants On Platelet Protease-Activated Receptor 1 ActivationDokumen6 halamanEffect of Aprotinin and Recombinant Variants On Platelet Protease-Activated Receptor 1 Activationfluid_man_brazilBelum ada peringkat
- Prevention of Postoperative Pericardial Adhesions With A Novel Regenerative Collagen SheetDokumen8 halamanPrevention of Postoperative Pericardial Adhesions With A Novel Regenerative Collagen Sheetfluid_man_brazilBelum ada peringkat
- Invited Commentary - Aprotinin and Recombinant Variants (Day Et Al)Dokumen1 halamanInvited Commentary - Aprotinin and Recombinant Variants (Day Et Al)fluid_man_brazilBelum ada peringkat
- Invited Commentary - Preoperative WBC Link To Cabg Outcome (Newall Et Al)Dokumen2 halamanInvited Commentary - Preoperative WBC Link To Cabg Outcome (Newall Et Al)fluid_man_brazilBelum ada peringkat
- Effect of Aprotinin and Recombinant Variants On Platelet Protease-Activated Receptor 1 ActivationDokumen6 halamanEffect of Aprotinin and Recombinant Variants On Platelet Protease-Activated Receptor 1 Activationfluid_man_brazilBelum ada peringkat
- Invited Commentary - No Volume-Outcome Relationship For Opcabg (Plomondon Et Al)Dokumen2 halamanInvited Commentary - No Volume-Outcome Relationship For Opcabg (Plomondon Et Al)fluid_man_brazilBelum ada peringkat
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (587)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (399)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2219)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (265)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (119)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- EPHI PHEOC COVID-19 Weekly Bulletin 24 English 10122020Dokumen16 halamanEPHI PHEOC COVID-19 Weekly Bulletin 24 English 10122020wubet tazebBelum ada peringkat
- Combating COVID-19 and Building Immune Resilience: A Potential Role For Magnesium Nutrition?Dokumen10 halamanCombating COVID-19 and Building Immune Resilience: A Potential Role For Magnesium Nutrition?witaBelum ada peringkat
- 2033 Rheumatoid ArthritisDokumen48 halaman2033 Rheumatoid Arthritisdinaarzina100% (1)
- ActemraDokumen7 halamanActemraLinda KvastadBelum ada peringkat
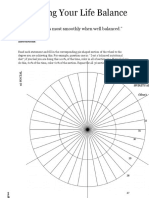
- Assessing Life Balance: A Concise Guide to Finding HarmonyDokumen17 halamanAssessing Life Balance: A Concise Guide to Finding HarmonyWiljohn de la CruzBelum ada peringkat
- Rheum at OlogyDokumen36 halamanRheum at OlogyCarlos HernándezBelum ada peringkat
- COVID-19 Management in Hospitalized Adults - UpToDateDokumen46 halamanCOVID-19 Management in Hospitalized Adults - UpToDateXochilt Mejia GarciaBelum ada peringkat
- Echinochrome A and Cytokine Storm SyndromeDokumen11 halamanEchinochrome A and Cytokine Storm SyndromeNicolas Fernandez RubilarBelum ada peringkat
- Tocilizumab CovidDokumen15 halamanTocilizumab CovidAponte MaríaBelum ada peringkat
- COVID 19 Science Report Therapeutics 4 May PDFDokumen41 halamanCOVID 19 Science Report Therapeutics 4 May PDFjolamo1122916Belum ada peringkat
- The Liver in Times of COVID-19 What Hepatologists Should KnowDokumen6 halamanThe Liver in Times of COVID-19 What Hepatologists Should KnowFita FitriantiBelum ada peringkat
- Model Biologics IndicationsDokumen25 halamanModel Biologics IndicationsVenkata Suryanarayana GorleBelum ada peringkat
- Tocilizumab For Treatment Patients With COVID-19 - Recommended Medication For Novel Disease.Dokumen8 halamanTocilizumab For Treatment Patients With COVID-19 - Recommended Medication For Novel Disease.Ginna Paola SaavedraBelum ada peringkat
- Cytokine Storm in COVID-19Dokumen11 halamanCytokine Storm in COVID-19Feli FelBelum ada peringkat
- Treatment of Graves' Orbitopathy (Ophthalmopathy) - UpToDateDokumen22 halamanTreatment of Graves' Orbitopathy (Ophthalmopathy) - UpToDateAli Aborges Jr.Belum ada peringkat
- Tocilizumab Drug Monograph - 1JUN2020Dokumen43 halamanTocilizumab Drug Monograph - 1JUN2020juanferreBelum ada peringkat
- Mallik Questions ReplyDokumen9 halamanMallik Questions ReplysanthoshBelum ada peringkat
- Medical Tribune January 2012 HKDokumen72 halamanMedical Tribune January 2012 HKKarena SabadoBelum ada peringkat
- Actemra PI May 2019Dokumen5 halamanActemra PI May 2019Fajar Sri Lestari PangukirBelum ada peringkat
- Viruses: Sars-Cov-2/Covid-19: Viral Genomics, Epidemiology, Vaccines, and Therapeutic InterventionsDokumen18 halamanViruses: Sars-Cov-2/Covid-19: Viral Genomics, Epidemiology, Vaccines, and Therapeutic InterventionsMahantesh NyayakarBelum ada peringkat
- Inform Concent Actemra 1Dokumen2 halamanInform Concent Actemra 1lukiharjantiBelum ada peringkat
- JAMA PMR and GCA ReviewDokumen2 halamanJAMA PMR and GCA ReviewFelicia.Octofinna IbrahimBelum ada peringkat
- Disease Modifying Anti-Rheumatic Drugs DMARDsDokumen8 halamanDisease Modifying Anti-Rheumatic Drugs DMARDsekoBelum ada peringkat
- A Review On Macrophage Activation SyndromeDokumen9 halamanA Review On Macrophage Activation Syndromeedy noveryBelum ada peringkat
- Possible treatments for COVID-19: review of remdesivir, dexamethasone and other agentsDokumen16 halamanPossible treatments for COVID-19: review of remdesivir, dexamethasone and other agentsAni RahayuBelum ada peringkat
- Uci Protocol Brighman BostonDokumen43 halamanUci Protocol Brighman BostonAnonymous ZUaUz1wwBelum ada peringkat
- TocilizumabDokumen14 halamanTocilizumabTri CahyaniBelum ada peringkat
- Rheumatoid Arthritis Treatment and Management GuideDokumen32 halamanRheumatoid Arthritis Treatment and Management GuideamirthaBelum ada peringkat
- Interstitial Lung Disease 2021 3Dokumen11 halamanInterstitial Lung Disease 2021 3Khanh Ha NguyenBelum ada peringkat
- Tocilizumab and Remdesivir in HospitalizedDokumen13 halamanTocilizumab and Remdesivir in HospitalizedYulian 53Belum ada peringkat