Anda mungkin juga menyukai
- Bash CookbookDokumen21 halamanBash CookbookReiko11Belum ada peringkat
- 389 Directory ServerDokumen24 halaman389 Directory ServerRajMohenBelum ada peringkat
- TAFJ DistributionDokumen11 halamanTAFJ DistributionShaqif Hasan SajibBelum ada peringkat
- Senior DBA Interview QuestionsDokumen8 halamanSenior DBA Interview QuestionsKadambari RanjanBelum ada peringkat
- Interview QuestionDokumen24 halamanInterview QuestionAnil YarlagaddaBelum ada peringkat
- Linux Interview QuestionsDokumen52 halamanLinux Interview Questionsvinnuthegreat100% (1)
- Troubleshooting LAVM10Dokumen40 halamanTroubleshooting LAVM10Txus NietoBelum ada peringkat
- 3JL01001BFAARJZZA - V1 - 5520 AMS Release 9.4 Installation and Migration GuideDokumen328 halaman3JL01001BFAARJZZA - V1 - 5520 AMS Release 9.4 Installation and Migration GuideMichał Buźniak100% (1)
- 11.3.1.1 Lab - Setup A Multi-VM EnvironmentDokumen7 halaman11.3.1.1 Lab - Setup A Multi-VM EnvironmentNoacto TwinrockBelum ada peringkat
- Build Your Own Distributed Compilation Cluster: A Practical WalkthroughDari EverandBuild Your Own Distributed Compilation Cluster: A Practical WalkthroughBelum ada peringkat
- Linux Interview QuestionsDokumen51 halamanLinux Interview Questionsseenu933Belum ada peringkat
- Apache Tomcat TuningDokumen20 halamanApache Tomcat Tuningapi-3784370100% (1)
- DBMS Complete Note PDFDokumen130 halamanDBMS Complete Note PDFSarwesh MaharzanBelum ada peringkat
- Volvo PTT 2.8 Install GuideDokumen10 halamanVolvo PTT 2.8 Install GuidePedro GomezBelum ada peringkat
- Oracle 11g RAC Implementation GuideDokumen38 halamanOracle 11g RAC Implementation Guidejatin_meghani100% (1)
- RAC 11gR2 CLUSTER SETUPDokumen82 halamanRAC 11gR2 CLUSTER SETUPvenkatreddyBelum ada peringkat
- Bigdata NotesDokumen26 halamanBigdata NotesAnil YarlagaddaBelum ada peringkat
- Installation Oracle RAC - AIXDokumen48 halamanInstallation Oracle RAC - AIXHung NguyenBelum ada peringkat
- Homer: Mapping Reads To The GenomeDokumen5 halamanHomer: Mapping Reads To The GenomecquintoBelum ada peringkat
- Image Detection With Lazarus: Maxbox Starter87 With CaiDokumen22 halamanImage Detection With Lazarus: Maxbox Starter87 With CaiMax Kleiner100% (1)
- AIX SambaPware PDFDokumen37 halamanAIX SambaPware PDFoliversusBelum ada peringkat
- In This Tutorial We Will Discuss How To Install KohaDokumen7 halamanIn This Tutorial We Will Discuss How To Install KohapaulordonezBelum ada peringkat
- UNIX-LINUX Shell CommandsDokumen10 halamanUNIX-LINUX Shell Commandskamy-gBelum ada peringkat
- Initializing A Build EnvironmentDokumen26 halamanInitializing A Build EnvironmentMuhammad AliBelum ada peringkat
- Brief IntroductionDokumen6 halamanBrief IntroductionKalyanakrishnan BalachandranBelum ada peringkat
- Arch Linux For DummiesDokumen28 halamanArch Linux For DummiestotosttsBelum ada peringkat
- WinionDokumen5 halamanWinionRolm BrosBelum ada peringkat
- How To Install Linux, Apache, Mysql, PHP (Lamp) Stack On Debian 9 StretchDokumen24 halamanHow To Install Linux, Apache, Mysql, PHP (Lamp) Stack On Debian 9 StretchHenry CahyoBelum ada peringkat
- WIEN2k InstallationDokumen12 halamanWIEN2k InstallationMohamed-Amine TalbiBelum ada peringkat
- WRF ArwDokumen8 halamanWRF Arwkeyur632100% (1)
- BkerndevDokumen38 halamanBkerndevekuleusBelum ada peringkat
- How To Cross Compile Python and Run in Embedded SystemDokumen12 halamanHow To Cross Compile Python and Run in Embedded Systemchuiyewleong100% (1)
- MIT6 858F14 Lab1Dokumen8 halamanMIT6 858F14 Lab1jarod_kyleBelum ada peringkat
- Using Ndisk64 NstressDokumen3 halamanUsing Ndisk64 NstressdanilaixBelum ada peringkat
- Denbi Metagenomics WorkshopDokumen21 halamanDenbi Metagenomics WorkshopJack SimBelum ada peringkat
- Mips Bof Lyonyang Public FinalDokumen28 halamanMips Bof Lyonyang Public FinalTan Phu NguyenBelum ada peringkat
- READMEDokumen3 halamanREADMEeifer4Belum ada peringkat
- Readme PDFDokumen19 halamanReadme PDFNhat MinhBelum ada peringkat
- How To Install MB-systemDokumen11 halamanHow To Install MB-systemAdenuga AdebisiBelum ada peringkat
- Gpfs ProblemsDokumen6 halamanGpfs ProblemsMq SfsBelum ada peringkat
- Modern Stack SmashingDokumen9 halamanModern Stack Smashinghacktivists2011Belum ada peringkat
- Static Functions in Linux Device DriverDokumen4 halamanStatic Functions in Linux Device DriverAkhilesh ChaudhryBelum ada peringkat
- WSTE KevinGrigorenko LinuxNativeDokumen60 halamanWSTE KevinGrigorenko LinuxNativedesaesBelum ada peringkat
- Emperor OS New Features v2Dokumen27 halamanEmperor OS New Features v2Saqib HussainBelum ada peringkat
- BDA LabCompendiumDokumen6 halamanBDA LabCompendiumKiS MintBelum ada peringkat
- Accessing The SystemDokumen32 halamanAccessing The SystemShivBelum ada peringkat
- What Is A Program?: Some Fundamentals (A Review)Dokumen18 halamanWhat Is A Program?: Some Fundamentals (A Review)Halil İbrahim ŞafakBelum ada peringkat
- What Is Cross Compilation - Cross Compilation DemystifiedDokumen10 halamanWhat Is Cross Compilation - Cross Compilation DemystifiedchuiyewleongBelum ada peringkat
- Benchmarking SAS® I/O: Verifying I/O Performance Using Fio: Spencer Hayes, DLL ConsultingDokumen7 halamanBenchmarking SAS® I/O: Verifying I/O Performance Using Fio: Spencer Hayes, DLL ConsultingUnduh BahanBelum ada peringkat
- 1053 - Inside Macosx Kernel PDFDokumen74 halaman1053 - Inside Macosx Kernel PDFErasmo Leite Jr.Belum ada peringkat
- Bytemeta - Vip Emefffcode Aster MPI in Singularity of SM2021Dokumen5 halamanBytemeta - Vip Emefffcode Aster MPI in Singularity of SM2021helloddBelum ada peringkat
- Work CoreDokumen5 halamanWork Corekhurs195Belum ada peringkat
- Word Count Exercise InstructionsDokumen3 halamanWord Count Exercise InstructionsAshwin AjmeraBelum ada peringkat
- Converting Your MyBook World Into A File and WebServerDokumen16 halamanConverting Your MyBook World Into A File and WebServerDavid de NieseBelum ada peringkat
- Lab - Locating Log Files: ObjectivesDokumen16 halamanLab - Locating Log Files: ObjectivesgeorgeBelum ada peringkat
- UnixDokumen5 halamanUnixIndrajeet Nigam100% (1)
- How To Configure Two Node High Availability Cluster On RHELDokumen18 halamanHow To Configure Two Node High Availability Cluster On RHELslides courseBelum ada peringkat
- Sun Grid Engine TutorialDokumen14 halamanSun Grid Engine TutorialoligoelementoBelum ada peringkat
- Brief History of Java-LectureDokumen10 halamanBrief History of Java-LectureMharbse EdzaBelum ada peringkat
- INTR C QnsDokumen41 halamanINTR C QnsMalli Naidu KarpurapuBelum ada peringkat
- Memory and ClassificationDokumen8 halamanMemory and Classificationsingh7863Belum ada peringkat
- Configuration of a Simple Samba File Server, Quota and Schedule BackupDari EverandConfiguration of a Simple Samba File Server, Quota and Schedule BackupBelum ada peringkat
- Configuration of a Simple Samba File Server, Quota and Schedule BackupDari EverandConfiguration of a Simple Samba File Server, Quota and Schedule BackupBelum ada peringkat
- Unit 1 Java FundamentalsDokumen51 halamanUnit 1 Java Fundamentalsanshulvyas23Belum ada peringkat
- Why Do We Study Theory of Computation ?Dokumen36 halamanWhy Do We Study Theory of Computation ?anshulvyas23Belum ada peringkat
- Exception Handling in JavaDokumen2 halamanException Handling in Javaanshulvyas23Belum ada peringkat
- MpiDokumen30 halamanMpianshulvyas23Belum ada peringkat
- M.Tech Final Year Project (Phase - 3) : Submited To Submited byDokumen23 halamanM.Tech Final Year Project (Phase - 3) : Submited To Submited byanshulvyas23Belum ada peringkat
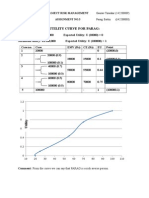
- Utility Curve For ParagDokumen2 halamanUtility Curve For Paraganshulvyas23Belum ada peringkat
- DPC Principal 2015 16 28042015Dokumen332 halamanDPC Principal 2015 16 28042015anshulvyas23Belum ada peringkat
- Practical Guide of Building A HPC ClusterDokumen30 halamanPractical Guide of Building A HPC ClusterPuteh SoloBelum ada peringkat
- Ms-Dos o Level 27-4-2020 PDFDokumen11 halamanMs-Dos o Level 27-4-2020 PDFPushpa kumariBelum ada peringkat
- Bridge Modeling I (OBM) ManualDokumen90 halamanBridge Modeling I (OBM) ManualSunilBelum ada peringkat
- How To Add Serial ATA (SATA) Drives To A Windows Installation - HP® SupportDokumen7 halamanHow To Add Serial ATA (SATA) Drives To A Windows Installation - HP® SupportiqtadarBelum ada peringkat
- Oracle 0212Dokumen80 halamanOracle 0212gamurphy65Belum ada peringkat
- KD-SR80BT: Instruction Manual Manual de Instrucciones Manuel D'InstructionsDokumen76 halamanKD-SR80BT: Instruction Manual Manual de Instrucciones Manuel D'InstructionsJulian DuarteBelum ada peringkat
- M4 SP Flash Tool Guidance V1.0 PDFDokumen12 halamanM4 SP Flash Tool Guidance V1.0 PDFkomander apodacaBelum ada peringkat
- Manual Works Prompter Reference V5.4Dokumen5 halamanManual Works Prompter Reference V5.4Emmanuel Nogales GomezBelum ada peringkat
- Openmediavault Readthedocs Io en 5.xDokumen167 halamanOpenmediavault Readthedocs Io en 5.xTony NGUYENBelum ada peringkat
- FS8600 Snapshot and Cloning Best PracticesDokumen27 halamanFS8600 Snapshot and Cloning Best PracticesManoj KarthiBelum ada peringkat
- CoreFramwork Developer ManualDokumen48 halamanCoreFramwork Developer ManualgudissagabissaBelum ada peringkat
- ANSYS BatchDokumen2 halamanANSYS BatchKumarBelum ada peringkat
- Operating System Concepts & Computer Fundamentals: Sachin G. Pawar Sunbeam, PuneDokumen100 halamanOperating System Concepts & Computer Fundamentals: Sachin G. Pawar Sunbeam, Punegolu patelBelum ada peringkat
- Aneka Installation GuideDokumen71 halamanAneka Installation GuideSathish KumarBelum ada peringkat
- MetroCount Data CollectionDokumen30 halamanMetroCount Data CollectionPaco TrooperBelum ada peringkat
- 70-698 Lesson07Dokumen23 halaman70-698 Lesson07Ramandeep kaurBelum ada peringkat
- GPR Import DisplayDokumen2 halamanGPR Import DisplayAlexandra GereaBelum ada peringkat
- Broadsoft Partner Configuration GuideDokumen46 halamanBroadsoft Partner Configuration GuideGabriel H. MartinezBelum ada peringkat
- Visionic 5.2 Release NotesDokumen2 halamanVisionic 5.2 Release NotesintorelBelum ada peringkat
- BA Configuration Tool Technical NoteDokumen4 halamanBA Configuration Tool Technical NotemauroboteroBelum ada peringkat
- ADOBEDokumen5 halamanADOBEFekadu ErkoBelum ada peringkat
- BelView NMS User Guide 5 3Dokumen285 halamanBelView NMS User Guide 5 3michael dejeneBelum ada peringkat
- Userguide v1.2Dokumen9 halamanUserguide v1.2Remus BulgaruBelum ada peringkat
- VIIRS Cloud Optical Properties Science Processing Algorithm (COP - SPA) User's GuideDokumen10 halamanVIIRS Cloud Optical Properties Science Processing Algorithm (COP - SPA) User's GuideNithindev GuttikondaBelum ada peringkat
- Kofax Web Capture: Developer's GuideDokumen194 halamanKofax Web Capture: Developer's GuideNicoleta FraticaBelum ada peringkat