Anda mungkin juga menyukai
- Anaphylactic Reactions in Anesthesia and Intensive CareDari EverandAnaphylactic Reactions in Anesthesia and Intensive CareBelum ada peringkat
- HHS Public Access: The Pathophysiology of AnaphylaxisDokumen29 halamanHHS Public Access: The Pathophysiology of AnaphylaxisdilaBelum ada peringkat
- Insights to Neuroimmune BiologyDari EverandInsights to Neuroimmune BiologyIstvan BercziBelum ada peringkat
- The Pathophysiology of Anaphylaxis: Mechanisms of Allergic DiseasesDokumen14 halamanThe Pathophysiology of Anaphylaxis: Mechanisms of Allergic DiseasesAprilihardini LaksmiBelum ada peringkat
- World Allergy OrganizationDokumen25 halamanWorld Allergy OrganizationHusnul HanifaBelum ada peringkat
- Tips - Anaphylaxis and Hypersensitivity Reactions PDFDokumen376 halamanTips - Anaphylaxis and Hypersensitivity Reactions PDFsorinproiecteBelum ada peringkat
- Johnson 2004Dokumen9 halamanJohnson 2004Abdul Wahab 2008126345Belum ada peringkat
- Update On Biphasic AnaphylaxisDokumen7 halamanUpdate On Biphasic AnaphylaxisTikaa RachmawatiBelum ada peringkat
- Anaphylaxis - 2Dokumen8 halamanAnaphylaxis - 2Ronald WiradirnataBelum ada peringkat
- Anaphylaxis: Review Open AccessDokumen7 halamanAnaphylaxis: Review Open AccessRadyaAgriBelum ada peringkat
- Seizures As Adverse Events of Antibiotic DrugsDokumen11 halamanSeizures As Adverse Events of Antibiotic DrugsCARZ 42Belum ada peringkat
- Anaphylaxis and AnesthesiaDokumen10 halamanAnaphylaxis and AnesthesiaKarlaBelum ada peringkat
- Vec 12066Dokumen18 halamanVec 12066Jose Efraím Olvera LopezBelum ada peringkat
- Anaphylaxis Diagnosis, TreatmentDokumen6 halamanAnaphylaxis Diagnosis, TreatmentutifornaBelum ada peringkat
- Epidemiology and Clinical Predictors of Biphasic Reactions in Children With AnaphylaxisDokumen9 halamanEpidemiology and Clinical Predictors of Biphasic Reactions in Children With AnaphylaxisPrihanant BayuBelum ada peringkat
- Nassal Provocation Test - 5Dokumen11 halamanNassal Provocation Test - 5Lengkung BajaBelum ada peringkat
- Alergic Desease PRACTALL-consensusDokumen12 halamanAlergic Desease PRACTALL-consensusNaty FernandezBelum ada peringkat
- Allergic Bronchopulmonary Aspergillosis: AAAAI Committee ReportDokumen6 halamanAllergic Bronchopulmonary Aspergillosis: AAAAI Committee ReportChristian Andrés CasasBelum ada peringkat
- Krishnaswamy 2021 - Critical Care Management of The Patient With Anaphylaxis - A Concise Definitive ReviewDokumen20 halamanKrishnaswamy 2021 - Critical Care Management of The Patient With Anaphylaxis - A Concise Definitive ReviewKhanh Ha NguyenBelum ada peringkat
- AnaphylaxisDokumen16 halamanAnaphylaxisEdgarBelum ada peringkat
- CDC 94616 DS1Dokumen15 halamanCDC 94616 DS1MadhuBelum ada peringkat
- Time of Onset and Predictors of Biphasic Anaphylactic Reactions: A Systematic Review and Meta-AnalysisDokumen11 halamanTime of Onset and Predictors of Biphasic Anaphylactic Reactions: A Systematic Review and Meta-AnalysisPrihanant BayuBelum ada peringkat
- The Management of Encephalitis Clinical Practice Guidelines by The Infectious Diseases Society of AmericaDokumen25 halamanThe Management of Encephalitis Clinical Practice Guidelines by The Infectious Diseases Society of AmericaNana ShkodinaBelum ada peringkat
- Drug Hypersensitivity: Pharmacogenetics and Clinical SyndromesDokumen7 halamanDrug Hypersensitivity: Pharmacogenetics and Clinical SyndromesdentsavvyBelum ada peringkat
- Autoimmune EncephalitisDokumen14 halamanAutoimmune EncephalitisMarco Antonio KoffBelum ada peringkat
- Schatz 2014Dokumen4 halamanSchatz 2014zavoianu.diana21Belum ada peringkat
- Anaphylactic Shock - Pathophysiology, Recognition, and Treatment PDFDokumen9 halamanAnaphylactic Shock - Pathophysiology, Recognition, and Treatment PDFTita Luthfia100% (1)
- Anaphylaxis in ChildrenDokumen8 halamanAnaphylaxis in Childrenapi-351303195Belum ada peringkat
- 412 FullDokumen14 halaman412 FullDennyBelum ada peringkat
- Peri-Operative AnaphylaxDokumen7 halamanPeri-Operative AnaphylaxKarlaBelum ada peringkat
- Antibiotic Associated EncephalopathyDokumen9 halamanAntibiotic Associated EncephalopathyAmila Atic JasarevicBelum ada peringkat
- HHS Public Access: Autoimmune EncephalopathiesDokumen33 halamanHHS Public Access: Autoimmune EncephalopathiesLink BuiBelum ada peringkat
- Estatus Epileptico de Nuevo Inicio (NORSE)Dokumen8 halamanEstatus Epileptico de Nuevo Inicio (NORSE)juanutBelum ada peringkat
- Voluntary Activation of The Sympathetic Nervous System and Attenuation of The Innate Immune Response in HumansDokumen6 halamanVoluntary Activation of The Sympathetic Nervous System and Attenuation of The Innate Immune Response in HumansGerson De La FuenteBelum ada peringkat
- Molecular Mechanisms in Genetically Defined Autoinflammatory Diseases. Disorders of Amplified Danger SignalingDokumen58 halamanMolecular Mechanisms in Genetically Defined Autoinflammatory Diseases. Disorders of Amplified Danger SignalingAlmudena MeiBelum ada peringkat
- Anaphylaxis: Learning ObjectivesDokumen6 halamanAnaphylaxis: Learning ObjectivesMentari FBelum ada peringkat
- Antioxidants 12 00280Dokumen62 halamanAntioxidants 12 00280emeo145Belum ada peringkat
- NIH Public Access: Insect Sting AnaphylaxisDokumen11 halamanNIH Public Access: Insect Sting AnaphylaxisdilaBelum ada peringkat
- IDSA Guidelines for Diagnosis and Treatment of EncephalitisDokumen25 halamanIDSA Guidelines for Diagnosis and Treatment of EncephalitisJizus GonzalezBelum ada peringkat
- Nassal Provocation Test - 6Dokumen5 halamanNassal Provocation Test - 6Lengkung BajaBelum ada peringkat
- An Update On Perioperative Anaphylaxis (11!04!2016)Dokumen6 halamanAn Update On Perioperative Anaphylaxis (11!04!2016)Maufer AlBelum ada peringkat
- Anafilaxia y Shock Anafiláctico: Anaphilactic Shock and AnaphilaxisDokumen29 halamanAnafilaxia y Shock Anafiláctico: Anaphilactic Shock and AnaphilaxissofiaBelum ada peringkat
- Alyu 2016Dokumen16 halamanAlyu 2016Jessy CABelum ada peringkat
- Impacto de La NoreDokumen8 halamanImpacto de La Noreyanie.jaeBelum ada peringkat
- Recommendations For Appropriate Sublingual Immunotherapy Clinical TrialsDokumen6 halamanRecommendations For Appropriate Sublingual Immunotherapy Clinical TrialstitatBelum ada peringkat
- Acupuncture ModulatesDokumen12 halamanAcupuncture ModulatesAdán LópezBelum ada peringkat
- Anaphylaxis: Diagnosis and Management: The Medical Journal of Australia October 2006Dokumen8 halamanAnaphylaxis: Diagnosis and Management: The Medical Journal of Australia October 2006DidiBelum ada peringkat
- The Management of Encephalitis: Clinical Practice Guidelines by The Infectious Diseases Society of AmericaDokumen25 halamanThe Management of Encephalitis: Clinical Practice Guidelines by The Infectious Diseases Society of AmericaVictor Euclides Briones MoralesBelum ada peringkat
- Autoimmune Encephalitis Review (Lancet 2016)Dokumen14 halamanAutoimmune Encephalitis Review (Lancet 2016)tjelongBelum ada peringkat
- Evidence-Based SLIT Benefits for Allergic RhinitisDokumen10 halamanEvidence-Based SLIT Benefits for Allergic RhinitisPeter SalimBelum ada peringkat
- Basophils and Allergic Inflammation: Mechanisms of Allergic DiseasesDokumen13 halamanBasophils and Allergic Inflammation: Mechanisms of Allergic DiseasesBasideu ByinajuBelum ada peringkat
- Autoimmune Encephalitis: Proposed Best Practice Recommendations For Diagnosis and Acute ManagementDokumen12 halamanAutoimmune Encephalitis: Proposed Best Practice Recommendations For Diagnosis and Acute ManagementSafitri MuhlisaBelum ada peringkat
- Encefalite Autoimune Melhores Praticas No Diagnostico e Manejo AgudoDokumen12 halamanEncefalite Autoimune Melhores Praticas No Diagnostico e Manejo AgudoAmandha AlencarBelum ada peringkat
- HHS Public Access: Antibiotic AllergyDokumen32 halamanHHS Public Access: Antibiotic AllergyInanda DamantiaBelum ada peringkat
- Infectious Dan Autoantibody Associated EncephalitisDokumen13 halamanInfectious Dan Autoantibody Associated EncephalitisAyudiah ParamitaBelum ada peringkat
- Vol28issue4 1Dokumen17 halamanVol28issue4 1BTS LGOBelum ada peringkat
- Epinephrine: The Drug of Choice For Anaphylaxisva Statement of The World Allergy OrganizationDokumen9 halamanEpinephrine: The Drug of Choice For Anaphylaxisva Statement of The World Allergy OrganizationnadiaaapudeBelum ada peringkat
- Myths, Facts and Controversies in The Diagnosis and Management of AnaphylaxisDokumen8 halamanMyths, Facts and Controversies in The Diagnosis and Management of AnaphylaxisWafaa AbdullahBelum ada peringkat
- 癫痫发病的分子机制Dokumen10 halaman癫痫发病的分子机制phuwinyangBelum ada peringkat
- MJR 28 2 64Dokumen6 halamanMJR 28 2 64jone6Belum ada peringkat
- Hi Scan Pro ManualDokumen231 halamanHi Scan Pro ManualFaridhul IkhsanBelum ada peringkat
- Aviation Case StudyDokumen6 halamanAviation Case Studynabil sayedBelum ada peringkat
- UntitledDokumen17 halamanUntitledВладислав ПроскураBelum ada peringkat
- DocuCentre IV C4470 3370 2270 BrochureDokumen8 halamanDocuCentre IV C4470 3370 2270 BrochureRumen StoychevBelum ada peringkat
- George F Kennan and The Birth of Containment The Greek Test CaseDokumen17 halamanGeorge F Kennan and The Birth of Containment The Greek Test CaseEllinikos Emfilios100% (1)
- Journal EntriesDokumen10 halamanJournal Entriesapi-283322366Belum ada peringkat
- Unit Revision-Integrated Systems For Business EnterprisesDokumen8 halamanUnit Revision-Integrated Systems For Business EnterprisesAbby JiangBelum ada peringkat
- Chapter 7 Project Cost ManagementDokumen48 halamanChapter 7 Project Cost Managementafifah suyadiBelum ada peringkat
- Ethical CRM PracticesDokumen21 halamanEthical CRM Practicesanon_522592057Belum ada peringkat
- Miranda V AgDokumen3 halamanMiranda V AgCARLO JOSE BACTOLBelum ada peringkat
- Cultural Practices and Academic Performance of Blaan Pupils in Sinapulan Elementary SchoolDokumen15 halamanCultural Practices and Academic Performance of Blaan Pupils in Sinapulan Elementary SchoolLorBelum ada peringkat
- SMAW Product DevelopmentDokumen9 halamanSMAW Product Developmenttibo bursioBelum ada peringkat
- LAWHIST - Week1 - Codamon Lim Tan PDFDokumen32 halamanLAWHIST - Week1 - Codamon Lim Tan PDFMargell TanBelum ada peringkat
- Cps InfographicDokumen1 halamanCps Infographicapi-665846419Belum ada peringkat
- The Philippine Army Doctrine DevelopmentDokumen10 halamanThe Philippine Army Doctrine DevelopmentRy PomarBelum ada peringkat
- Review Unit 10 Test CHP 17Dokumen13 halamanReview Unit 10 Test CHP 17TechnoKittyKittyBelum ada peringkat
- Cartha Worth SharingDokumen27 halamanCartha Worth SharingtereAC85Belum ada peringkat
- High Intermediate Analogies 9Dokumen2 halamanHigh Intermediate Analogies 9Usman KhalidBelum ada peringkat
- Sample File: Official Game AccessoryDokumen6 halamanSample File: Official Game AccessoryJose L GarcíaBelum ada peringkat
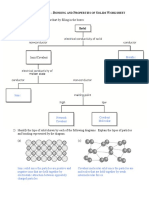
- Bonding and Properties of Solids Worksheet Solutions 1kadax6Dokumen4 halamanBonding and Properties of Solids Worksheet Solutions 1kadax6Mel Patricia M. CabreraBelum ada peringkat
- Para Kay BDokumen1 halamanPara Kay BFeLy DipOn63% (8)
- Red ProjectDokumen30 halamanRed ProjectApoorva SrivastavaBelum ada peringkat
- IS-LM Model Analysis of Monetary and Fiscal PolicyDokumen23 halamanIS-LM Model Analysis of Monetary and Fiscal PolicyFatima mirzaBelum ada peringkat
- Edukasyon Sa Pagpapakatao (Esp) Monitoring and Evaluation Tool For Department Heads/Chairmen/CoordinatorsDokumen3 halamanEdukasyon Sa Pagpapakatao (Esp) Monitoring and Evaluation Tool For Department Heads/Chairmen/CoordinatorsPrincis CianoBelum ada peringkat
- Enterprise Information Management (EIM) : by Katlego LeballoDokumen9 halamanEnterprise Information Management (EIM) : by Katlego LeballoKatlego LeballoBelum ada peringkat
- Asian Paints Research ProposalDokumen1 halamanAsian Paints Research ProposalYASH JOHRI-DM 21DM222Belum ada peringkat
- Chapter 1 Introduction To Management and OrganisationDokumen34 halamanChapter 1 Introduction To Management and Organisationsahil malhotraBelum ada peringkat
- 04-DDD.Assignment 2 frontsheet 2018-2019-đã chuyển đổi PDFDokumen21 halaman04-DDD.Assignment 2 frontsheet 2018-2019-đã chuyển đổi PDFl1111c1anh-5Belum ada peringkat
- Practical and Mathematical Skills BookletDokumen30 halamanPractical and Mathematical Skills BookletZarqaYasminBelum ada peringkat
- Hem Tiwari Vs Nidhi Tiwari Mutual Divorce - Revised VersionDokumen33 halamanHem Tiwari Vs Nidhi Tiwari Mutual Divorce - Revised VersionKesar Singh SawhneyBelum ada peringkat
- Crypt: Life, Death and Disease in the Middle Ages and BeyondDari EverandCrypt: Life, Death and Disease in the Middle Ages and BeyondPenilaian: 4 dari 5 bintang4/5 (3)
- Mitochondria and the Future of Medicine: The Key to Understanding Disease, Chronic Illness, Aging, and Life ItselfDari EverandMitochondria and the Future of Medicine: The Key to Understanding Disease, Chronic Illness, Aging, and Life ItselfPenilaian: 4.5 dari 5 bintang4.5/5 (98)
- Why We Die: The New Science of Aging and the Quest for ImmortalityDari EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityPenilaian: 3.5 dari 5 bintang3.5/5 (2)
- This Is Your Brain On Parasites: How Tiny Creatures Manipulate Our Behavior and Shape SocietyDari EverandThis Is Your Brain On Parasites: How Tiny Creatures Manipulate Our Behavior and Shape SocietyPenilaian: 3.5 dari 5 bintang3.5/5 (31)
- The Mind & The Brain: Neuroplasticity and the Power of Mental ForceDari EverandThe Mind & The Brain: Neuroplasticity and the Power of Mental ForceBelum ada peringkat
- 10% Human: How Your Body's Microbes Hold the Key to Health and HappinessDari Everand10% Human: How Your Body's Microbes Hold the Key to Health and HappinessPenilaian: 4 dari 5 bintang4/5 (33)
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisDari EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisPenilaian: 3.5 dari 5 bintang3.5/5 (2)
- Wayfinding: The Science and Mystery of How Humans Navigate the WorldDari EverandWayfinding: The Science and Mystery of How Humans Navigate the WorldPenilaian: 4.5 dari 5 bintang4.5/5 (18)
- All That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesDari EverandAll That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesPenilaian: 4.5 dari 5 bintang4.5/5 (396)
- The Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindDari EverandThe Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindPenilaian: 4.5 dari 5 bintang4.5/5 (93)
- Gathering Moss: A Natural and Cultural History of MossesDari EverandGathering Moss: A Natural and Cultural History of MossesPenilaian: 4.5 dari 5 bintang4.5/5 (347)
- Masterminds: Genius, DNA, and the Quest to Rewrite LifeDari EverandMasterminds: Genius, DNA, and the Quest to Rewrite LifeBelum ada peringkat
- The Lives of Bees: The Untold Story of the Honey Bee in the WildDari EverandThe Lives of Bees: The Untold Story of the Honey Bee in the WildPenilaian: 4.5 dari 5 bintang4.5/5 (44)
- The Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceDari EverandThe Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RacePenilaian: 4.5 dari 5 bintang4.5/5 (515)
- A Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsDari EverandA Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsPenilaian: 4.5 dari 5 bintang4.5/5 (4)
- Superlative: The Biology of ExtremesDari EverandSuperlative: The Biology of ExtremesPenilaian: 4.5 dari 5 bintang4.5/5 (51)
- The Confident Mind: A Battle-Tested Guide to Unshakable PerformanceDari EverandThe Confident Mind: A Battle-Tested Guide to Unshakable PerformancePenilaian: 4.5 dari 5 bintang4.5/5 (45)
- Why We Sleep: Unlocking the Power of Sleep and DreamsDari EverandWhy We Sleep: Unlocking the Power of Sleep and DreamsPenilaian: 4.5 dari 5 bintang4.5/5 (2083)
- The Dragons of Eden: Speculations on the Evolution of Human IntelligenceDari EverandThe Dragons of Eden: Speculations on the Evolution of Human IntelligencePenilaian: 4 dari 5 bintang4/5 (632)
- The Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorDari EverandThe Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorBelum ada peringkat
- Human Errors: A Panorama of Our Glitches, from Pointless Bones to Broken GenesDari EverandHuman Errors: A Panorama of Our Glitches, from Pointless Bones to Broken GenesPenilaian: 3.5 dari 5 bintang3.5/5 (55)
- Fearfully and Wonderfully: The Marvel of Bearing God's ImageDari EverandFearfully and Wonderfully: The Marvel of Bearing God's ImagePenilaian: 5 dari 5 bintang5/5 (40)
- Unthinkable: An Extraordinary Journey Through the World's Strangest BrainsDari EverandUnthinkable: An Extraordinary Journey Through the World's Strangest BrainsBelum ada peringkat
- Awkward: The Science of Why We're Socially Awkward and Why That's AwesomeDari EverandAwkward: The Science of Why We're Socially Awkward and Why That's AwesomePenilaian: 4 dari 5 bintang4/5 (23)
- Minds Make Societies: How Cognition Explains the World Humans CreateDari EverandMinds Make Societies: How Cognition Explains the World Humans CreatePenilaian: 4.5 dari 5 bintang4.5/5 (23)
- Inside of a Dog: What Dogs See, Smell, and KnowDari EverandInside of a Dog: What Dogs See, Smell, and KnowPenilaian: 4 dari 5 bintang4/5 (390)
- The Nature Fix: Why Nature Makes us Happier, Healthier, and More CreativeDari EverandThe Nature Fix: Why Nature Makes us Happier, Healthier, and More CreativePenilaian: 4 dari 5 bintang4/5 (157)
- The Dog Who Couldn't Stop Loving: How Dogs Have Captured Our Hearts for Thousands of YearsDari EverandThe Dog Who Couldn't Stop Loving: How Dogs Have Captured Our Hearts for Thousands of YearsBelum ada peringkat