Anda mungkin juga menyukai
- Fiber Dissection Technique: Lateral Aspect of The Brain: Surgical Anatomy and TechniqueDokumen11 halamanFiber Dissection Technique: Lateral Aspect of The Brain: Surgical Anatomy and TechniqueRafaelBelum ada peringkat
- Draft Guideline Clinical Investigation Medicinal Products Treatment Prevention Diabetes Mellitus - enDokumen24 halamanDraft Guideline Clinical Investigation Medicinal Products Treatment Prevention Diabetes Mellitus - enRafaelBelum ada peringkat
- Servello2008Dokumen8 halamanServello2008RafaelBelum ada peringkat
- Vestigation Medicinal Products Treatment Prevention Diabetes Mellitus Revision - enDokumen28 halamanVestigation Medicinal Products Treatment Prevention Diabetes Mellitus Revision - enRafaelBelum ada peringkat
- Porta2009Dokumen8 halamanPorta2009RafaelBelum ada peringkat
- UntitledDokumen316 halamanUntitledRafaelBelum ada peringkat
- LMOSMOOSMMOAMINISNKDokumen16 halamanLMOSMOOSMMOAMINISNKRafaelBelum ada peringkat
- Welter2008Dokumen6 halamanWelter2008RafaelBelum ada peringkat
- Martinez-Fernandez 2011Dokumen9 halamanMartinez-Fernandez 2011RafaelBelum ada peringkat
- Intra and Extradural Anterior Clinoidectomy: Anatomy Review and Surgical Technique Step by StepDokumen13 halamanIntra and Extradural Anterior Clinoidectomy: Anatomy Review and Surgical Technique Step by StepRafaelBelum ada peringkat
- Focus: Focused Ultrasound-Mediated Noninvasive Blood-BrainDokumen10 halamanFocus: Focused Ultrasound-Mediated Noninvasive Blood-BrainRafaelBelum ada peringkat
- Focus: NeurosurgicalDokumen10 halamanFocus: NeurosurgicalRafaelBelum ada peringkat
- FocusDokumen7 halamanFocusRafaelBelum ada peringkat
- Focus: Predicting Lesion Size During Focused Ultrasound Thalamotomy: A Review of 63 Lesions Over 3 Clinical TrialsDokumen6 halamanFocus: Predicting Lesion Size During Focused Ultrasound Thalamotomy: A Review of 63 Lesions Over 3 Clinical TrialsRafaelBelum ada peringkat
- Focus: Tremor, Thalamotomy, and CognitionDokumen2 halamanFocus: Tremor, Thalamotomy, and CognitionRafaelBelum ada peringkat
- FIPAT TA2 Part 4Dokumen69 halamanFIPAT TA2 Part 4Selim B. ÇerkeziBelum ada peringkat
- HdhdudhdhDokumen20 halamanHdhdudhdhRafaelBelum ada peringkat
- Chagas Disease: SeminarDokumen13 halamanChagas Disease: SeminarRafaelBelum ada peringkat
- Anterior Circulation Aneurysms: Clinic Al PearlsDokumen19 halamanAnterior Circulation Aneurysms: Clinic Al PearlsRafaelBelum ada peringkat
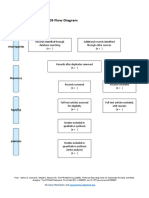
- PRISMA 2009 Flow Diagram GuideDokumen1 halamanPRISMA 2009 Flow Diagram GuideKou YashiroBelum ada peringkat
- Buffett's Alpha: Andrea Frazzini, David Kabiller, and Lasse Heje PedersenDokumen34 halamanBuffett's Alpha: Andrea Frazzini, David Kabiller, and Lasse Heje PedersenRafaelBelum ada peringkat
- TvyvtcrcececrcDokumen5 halamanTvyvtcrcececrcRafaelBelum ada peringkat
- Surgical Anatomy of The Jugular Foramen PDFDokumen9 halamanSurgical Anatomy of The Jugular Foramen PDFRafaelBelum ada peringkat
- Pharmacomechanical Catheter-Directed Thrombolysis For Deep-Vein ThrombosisDokumen13 halamanPharmacomechanical Catheter-Directed Thrombolysis For Deep-Vein ThrombosisRafaelBelum ada peringkat
- Acute Spinal Cord CompressionDokumen12 halamanAcute Spinal Cord CompressionRoberto López Mata100% (1)
- Anterior Circulation Aneurysms: Clinic Al PearlsDokumen19 halamanAnterior Circulation Aneurysms: Clinic Al PearlsRafaelBelum ada peringkat
- Pharmacomechanical Catheter-Directed Thrombolysis For Deep-Vein ThrombosisDokumen13 halamanPharmacomechanical Catheter-Directed Thrombolysis For Deep-Vein ThrombosisRafaelBelum ada peringkat
- Papel Dos Biologicos Jaci Pract Jan 2017Dokumen14 halamanPapel Dos Biologicos Jaci Pract Jan 2017RafaelBelum ada peringkat
- DjlnknekjecDokumen7 halamanDjlnknekjecRafaelBelum ada peringkat
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (894)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (399)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (265)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2219)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (119)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- Eaclipt Cpe 2019Dokumen9 halamanEaclipt Cpe 2019inas zahraBelum ada peringkat
- Hemoroid Interna Grade IV: By: Khairunnis A G1A21801 1Dokumen34 halamanHemoroid Interna Grade IV: By: Khairunnis A G1A21801 1Bella ReynaldiBelum ada peringkat
- Red EyesDokumen3 halamanRed EyesirijoaBelum ada peringkat
- Asa Physical Status Classification System: By: Dr. Ali Anwar SutisnaDokumen8 halamanAsa Physical Status Classification System: By: Dr. Ali Anwar Sutisnawan rennyBelum ada peringkat
- Ferenczi - My Friendship With Miksa SchachterDokumen4 halamanFerenczi - My Friendship With Miksa SchachtereuaggBelum ada peringkat
- The Tuskegee Syphilis StudyDokumen11 halamanThe Tuskegee Syphilis Studyapi-285171544Belum ada peringkat
- Euro J of Neurology - 2021 - Van Den Bergh - European Academy of Neurology Peripheral Nerve Society Guideline On DiagnosisDokumen28 halamanEuro J of Neurology - 2021 - Van Den Bergh - European Academy of Neurology Peripheral Nerve Society Guideline On DiagnosisSusana RocheBelum ada peringkat
- Chronic Limb IschemiaDokumen29 halamanChronic Limb IschemiaSadia NaveedBelum ada peringkat
- Mitek Anchor SurgeryDokumen9 halamanMitek Anchor SurgeryHeidy IxcaraguaBelum ada peringkat
- COE WorksheetDokumen16 halamanCOE WorksheetiloveraynaBelum ada peringkat
- Critiquing 1Dokumen3 halamanCritiquing 1Nilesh VeerBelum ada peringkat
- Nerve Conduction StudiesDokumen5 halamanNerve Conduction Studiessen ABelum ada peringkat
- Anc 0001 0009 0151Dokumen42 halamanAnc 0001 0009 0151ManhaBelum ada peringkat
- Nausea and VomitingDokumen9 halamanNausea and VomitingHusaifah BaliwanBelum ada peringkat
- LeprosyDokumen7 halamanLeprosytankmpBelum ada peringkat
- Chemotherapeutic Agents and AntiinfectivesDokumen64 halamanChemotherapeutic Agents and AntiinfectivesAnthony RiggsBelum ada peringkat
- Yakult ProfileDokumen36 halamanYakult ProfileMubeen NavazBelum ada peringkat
- Who Trs 993 Web FinalDokumen284 halamanWho Trs 993 Web FinalAnonymous 6OPLC9UBelum ada peringkat
- Memory Reconsolidation: Updating Memories Through RetrievalDokumen23 halamanMemory Reconsolidation: Updating Memories Through RetrievalRoberto LiaskowskyBelum ada peringkat
- Dramatizing Dementia: Madness in The Plays of Tennessee WilliamsDokumen8 halamanDramatizing Dementia: Madness in The Plays of Tennessee WilliamsMarcusFelsmanBelum ada peringkat
- FurosemideDokumen4 halamanFurosemideapi-3797941100% (1)
- Professional Development of Obstetrician & GynaecologistDokumen116 halamanProfessional Development of Obstetrician & GynaecologistDeepa YadavBelum ada peringkat
- Congenital Syphilis Causes and TreatmentDokumen4 halamanCongenital Syphilis Causes and TreatmentAlya Putri Khairani100% (1)
- Immunology & Oncology Review 2Dokumen99 halamanImmunology & Oncology Review 2Melchor Felipe SalvosaBelum ada peringkat
- Dim Mak and Acupuncture: Understanding the Medical Reasons Behind this Martial ArtDokumen6 halamanDim Mak and Acupuncture: Understanding the Medical Reasons Behind this Martial ArtIoan NeacsuBelum ada peringkat
- Safety Data Sheet for Masterseal 345Dokumen7 halamanSafety Data Sheet for Masterseal 345mkashkooli_scribdBelum ada peringkat
- CBT For CandA With OCD PDFDokumen16 halamanCBT For CandA With OCD PDFRoxana AlexandruBelum ada peringkat
- Sociology Final ProjectDokumen16 halamanSociology Final Projectabt09Belum ada peringkat
- A Review of The Use of The Health Belief Model For Weight ManagementDokumen1 halamanA Review of The Use of The Health Belief Model For Weight ManagementpatresyaBelum ada peringkat
- Supplemental Appendix 1Dokumen2 halamanSupplemental Appendix 1Ana Carolina SouzaBelum ada peringkat