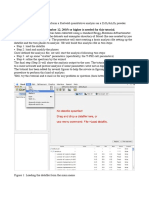
Anda mungkin juga menyukai
- Varian NMRDokumen4 halamanVarian NMRbram.soenen1Belum ada peringkat
- AutoCAD 2010 Tutorial Series: Drawing Dimensions, Elevations and SectionsDari EverandAutoCAD 2010 Tutorial Series: Drawing Dimensions, Elevations and SectionsBelum ada peringkat
- Mat152 Lab 4Dokumen6 halamanMat152 Lab 4Vishal NaikBelum ada peringkat
- M Nova Quick GuideDokumen6 halamanM Nova Quick GuideALI MUHAMMADBelum ada peringkat
- The Practically Cheating Statistics Handbook TI-83 Companion GuideDari EverandThe Practically Cheating Statistics Handbook TI-83 Companion GuidePenilaian: 3.5 dari 5 bintang3.5/5 (3)
- Dipole - Hfss 1Dokumen15 halamanDipole - Hfss 1shahnaz1981fatBelum ada peringkat
- Directions (Convolution Calculator v1.00)Dokumen4 halamanDirections (Convolution Calculator v1.00)anuragt_5Belum ada peringkat
- Power Transmission Line SimulationDokumen9 halamanPower Transmission Line SimulationArun B ThahaBelum ada peringkat
- Tutorial: How To Evaluate A Raman Map With Origin Pro 9.0Dokumen7 halamanTutorial: How To Evaluate A Raman Map With Origin Pro 9.0nselvakumar_254561Belum ada peringkat
- MestReNova 2DDokumen4 halamanMestReNova 2DMauricio Acelas M.Belum ada peringkat
- PSpice16 6 TutorialDokumen20 halamanPSpice16 6 TutorialAmbili MuraliBelum ada peringkat
- PSpice Tutorial Orcad 10Dokumen29 halamanPSpice Tutorial Orcad 10love_anish4274Belum ada peringkat
- X Bar&RchartDokumen9 halamanX Bar&RchartPramod AthiyarathuBelum ada peringkat
- Interpreting DSC Data V1aDokumen12 halamanInterpreting DSC Data V1aAzhariBelum ada peringkat
- 5 2021 11 0911 19 47 AmDokumen25 halaman5 2021 11 0911 19 47 Am29 - 003 - MD.Obayed HasanBelum ada peringkat
- HFSS - The Rectangular Patch AntennaDokumen16 halamanHFSS - The Rectangular Patch AntennaIvan ĐỗBelum ada peringkat
- RECON Getting StartedDokumen26 halamanRECON Getting Startednazmul hasanBelum ada peringkat
- 1 - Introduction To OrcadDokumen4 halaman1 - Introduction To OrcadRoxana RazecBelum ada peringkat
- Guide To NMR Data File Access and Mestrenova (Mnova) Data ProcessingDokumen3 halamanGuide To NMR Data File Access and Mestrenova (Mnova) Data ProcessingAndrew LondonBelum ada peringkat
- All Three Programs Indexing Result: Peaklist1Dokumen2 halamanAll Three Programs Indexing Result: Peaklist1Jeyadheepan KaBelum ada peringkat
- Introduction To ORCADDokumen15 halamanIntroduction To ORCADCruise_IceBelum ada peringkat
- AssignmentDokumen88 halamanAssignmentAsaf KhanBelum ada peringkat
- Cantilever Beam Example: Preprocessing: Defining The ProblemDokumen8 halamanCantilever Beam Example: Preprocessing: Defining The Problemabdul kareeBelum ada peringkat
- Convolutional Neural Networks (LeNet) - DeepLearning 0.1 DocumentationDokumen12 halamanConvolutional Neural Networks (LeNet) - DeepLearning 0.1 DocumentationSumit PatelBelum ada peringkat
- Getting Started 2011Dokumen5 halamanGetting Started 2011Edgar PuenteBelum ada peringkat
- Experiment 1 Half-Wave Dipole: RD STDokumen6 halamanExperiment 1 Half-Wave Dipole: RD STAntónio Domingos CongoBelum ada peringkat
- QPA TrainingDokumen13 halamanQPA TrainingsenthilkumarBelum ada peringkat
- Emeraude v5.20 - Doc v5.20.03 © KAPPA 1988-2019 PL Tutorial #1 - PLEX01 - 1/26Dokumen26 halamanEmeraude v5.20 - Doc v5.20.03 © KAPPA 1988-2019 PL Tutorial #1 - PLEX01 - 1/26carlos schoepsBelum ada peringkat
- Instructions For Using WinplotDokumen38 halamanInstructions For Using WinplotClaudia MuñozBelum ada peringkat
- Problem 3Dokumen37 halamanProblem 3Fredy Martin Humpiri ArelaBelum ada peringkat
- 02 Surface Water Hydrologymetric 2014fDokumen36 halaman02 Surface Water Hydrologymetric 2014fAngel Ariel Campos MurguiaBelum ada peringkat
- TP1 Intro2020 enDokumen7 halamanTP1 Intro2020 enbrunogassogbaBelum ada peringkat
- Manual Prisa 1.0Dokumen7 halamanManual Prisa 1.0Anisse CHIALIBelum ada peringkat
- How To Use TNavigatorDokumen55 halamanHow To Use TNavigatorMuhammadMulyawan100% (1)
- An Xmgrace TutorialDokumen8 halamanAn Xmgrace TutorialRen-Bo WangBelum ada peringkat
- Linear and Non Linear On PspiceDokumen11 halamanLinear and Non Linear On PspiceAseem GhimireBelum ada peringkat
- Lab 4. Point Pattern AnalysisDokumen38 halamanLab 4. Point Pattern AnalysisDaniyal AbbasiBelum ada peringkat
- RadShield Documentation v1.2Dokumen36 halamanRadShield Documentation v1.2fara latifaBelum ada peringkat
- Lab 1: Introduction To Pspice: ObjectivesDokumen6 halamanLab 1: Introduction To Pspice: ObjectivesJajagshshhshhBelum ada peringkat
- Radshield NCRP Example: Radiographic Room: Getting StartedDokumen33 halamanRadshield NCRP Example: Radiographic Room: Getting StartedDuong DucBelum ada peringkat
- KAPPA Rubis TutorialDokumen22 halamanKAPPA Rubis TutorialSARTHAK BAPAT100% (1)
- Ansys Tutorial For Lamb Waves PropagationDokumen12 halamanAnsys Tutorial For Lamb Waves PropagationRamy100% (1)
- Geo Soft TipsDokumen7 halamanGeo Soft TipsMuhammad Nanda ZyBelum ada peringkat
- User Manual EnglishDokumen30 halamanUser Manual EnglishmrcamupBelum ada peringkat
- Fluent Tutorial 1 - Fluid Flow and Heat Transfer in A Mixing ElbowDokumen56 halamanFluent Tutorial 1 - Fluid Flow and Heat Transfer in A Mixing Elbowklausosho100% (1)
- Shimadzu UV-1800 InstructionsDokumen5 halamanShimadzu UV-1800 InstructionsMarlon Mejia GuzmanBelum ada peringkat
- Bruker: 2D NMR Spectroscopy OverviewDokumen8 halamanBruker: 2D NMR Spectroscopy OverviewrenatoporangaBelum ada peringkat
- Erdas 1: Introduction To Erdas Imagine Goals:: Lanier - ImgDokumen8 halamanErdas 1: Introduction To Erdas Imagine Goals:: Lanier - ImgRêber MohammedBelum ada peringkat
- Tutorial RubisDokumen22 halamanTutorial RubisTeban CastroBelum ada peringkat
- Introduction To Pspice PDFDokumen7 halamanIntroduction To Pspice PDFAseem GhimireBelum ada peringkat
- Module XLC - Additional Information On Graphs in EXCEL Learning ObjectivesDokumen13 halamanModule XLC - Additional Information On Graphs in EXCEL Learning ObjectivesM VetriselviBelum ada peringkat
- Ansoft HFSS Version 8 / 8.5 Training Workbook: Slot AntennaDokumen23 halamanAnsoft HFSS Version 8 / 8.5 Training Workbook: Slot AntennaNurul Fahmi AriefBelum ada peringkat
- RSM Part 1Dokumen53 halamanRSM Part 1chirag chawareBelum ada peringkat
- Summer InternshipDokumen74 halamanSummer InternshipAatish kumar0% (1)
- XPSWMM Metric Tutorial 2Dokumen36 halamanXPSWMM Metric Tutorial 2Diego Sebastián Castillo PérezBelum ada peringkat
- Orcad 9.2 Lite Edition Getting Started GuideDokumen7 halamanOrcad 9.2 Lite Edition Getting Started GuideCloud WindBelum ada peringkat
- Set Xtics Font "Times-Roman, 30"Dokumen5 halamanSet Xtics Font "Times-Roman, 30"Chuck_YoungBelum ada peringkat
- Depth Map ManualDokumen4 halamanDepth Map ManualJoost de BontBelum ada peringkat
- K15 Population AveragesDokumen5 halamanK15 Population AveragesRigel_TBelum ada peringkat
- Religion, Ethnicity and Contested Nationhood in The Former Ottoman EmpireDokumen303 halamanReligion, Ethnicity and Contested Nationhood in The Former Ottoman EmpireRigel_T100% (2)
- Overview CurriculumDokumen1 halamanOverview CurriculumRigel_TBelum ada peringkat
- Nabuurs Et Al. Concepts Magn. Reson. 22A 90-105 2004Dokumen16 halamanNabuurs Et Al. Concepts Magn. Reson. 22A 90-105 2004Rigel_TBelum ada peringkat
- Racial Idea in The Independent State of CroatiaDokumen254 halamanRacial Idea in The Independent State of CroatiaRigel_TBelum ada peringkat
- Practical Manual: General Outline To Use The Structural Information Obtained From Molecular AlignmentDokumen19 halamanPractical Manual: General Outline To Use The Structural Information Obtained From Molecular AlignmentRigel_TBelum ada peringkat
- The Islamisation of The Meglen Vlachs (Megleno-Romanians) : The Village of Na Nti (No Tia) and The "Na Ntinets" in Present-Day TurkeyDokumen21 halamanThe Islamisation of The Meglen Vlachs (Megleno-Romanians) : The Village of Na Nti (No Tia) and The "Na Ntinets" in Present-Day TurkeyRigel_TBelum ada peringkat
- Molecular Modelling in Structure Determination: Structure Calculation From NMR DataDokumen13 halamanMolecular Modelling in Structure Determination: Structure Calculation From NMR DataRigel_TBelum ada peringkat
- P1&P2Dokumen32 halamanP1&P2Rigel_TBelum ada peringkat
- Practical RelaxDokumen5 halamanPractical RelaxRigel_TBelum ada peringkat
- Introductory Remarks VsDokumen4 halamanIntroductory Remarks VsRigel_TBelum ada peringkat
- De Alba&TjandraDokumen23 halamanDe Alba&TjandraRigel_TBelum ada peringkat
- NMR Past Future 2013Dokumen54 halamanNMR Past Future 2013Rigel_TBelum ada peringkat
- Sparky: Graphical NMR Assignment and Integration Program For Proteins, Nucleic Acids, and Other PolymersDokumen12 halamanSparky: Graphical NMR Assignment and Integration Program For Proteins, Nucleic Acids, and Other PolymersRigel_TBelum ada peringkat
- EMBO 2013 HandoutDeuterationDokumen54 halamanEMBO 2013 HandoutDeuterationRigel_TBelum ada peringkat
- Programme EMBO Course NMR Basel 2013: Meet Instructors and Tutors With Your Data and QuestionsDokumen4 halamanProgramme EMBO Course NMR Basel 2013: Meet Instructors and Tutors With Your Data and QuestionsRigel_TBelum ada peringkat
- EMBO Practical NMR Course 2013Dokumen6 halamanEMBO Practical NMR Course 2013Rigel_TBelum ada peringkat
- Jonker Rna Structure 2013Dokumen19 halamanJonker Rna Structure 2013Rigel_TBelum ada peringkat
- Wenmr: Introduction With TALOS+ and CYANA PracticalDokumen12 halamanWenmr: Introduction With TALOS+ and CYANA PracticalRigel_TBelum ada peringkat
- EMBO Practical Course July 20-27, 2013 P15: Building An HNCO From ScratchDokumen26 halamanEMBO Practical Course July 20-27, 2013 P15: Building An HNCO From ScratchRigel_TBelum ada peringkat
- P7 RDC RelaxDokumen30 halamanP7 RDC RelaxRigel_TBelum ada peringkat
- Grzesiek Relaxation TextDokumen37 halamanGrzesiek Relaxation TextRigel_TBelum ada peringkat
- Trosy Basel 2013aDokumen109 halamanTrosy Basel 2013aRigel_TBelum ada peringkat
- Itc Current ProtocolsDokumen24 halamanItc Current ProtocolsRigel_TBelum ada peringkat
- Designing Itc v7 ExptsDokumen5 halamanDesigning Itc v7 ExptsRigel_TBelum ada peringkat
- Swann Security System User Manual M89 - 4200H181113E - WebDokumen62 halamanSwann Security System User Manual M89 - 4200H181113E - WebOak CrestBelum ada peringkat
- Synergy PDFDokumen18 halamanSynergy PDFOliverBelum ada peringkat
- Wood Express ManualDokumen36 halamanWood Express ManualgertjaniBelum ada peringkat
- Manual Software para GillDokumen34 halamanManual Software para GillsoldadouniverBelum ada peringkat
- Logix Pro All LabsDokumen78 halamanLogix Pro All LabsJonathan Damn Dawn100% (3)
- Fluid SimDokumen387 halamanFluid SimArmando Campos Salazar100% (2)
- INTELLISPEC SERIE V-Páginas-31-46Dokumen16 halamanINTELLISPEC SERIE V-Páginas-31-46Antonio Valencia VillejoBelum ada peringkat
- Module 1 CC 101Dokumen33 halamanModule 1 CC 101Piyo100% (1)
- EEE-463 Antenna and Radio Wave Propagation: ComsatsDokumen27 halamanEEE-463 Antenna and Radio Wave Propagation: ComsatsMaaz Bin RizwanBelum ada peringkat
- Split Desktop ManualDokumen116 halamanSplit Desktop ManualAlexia Bernal100% (3)
- IT Skill LabDokumen37 halamanIT Skill LabSeven Lyx100% (1)
- User ManualDokumen90 halamanUser ManualРоманBelum ada peringkat
- Computer Hardware SummaryDokumen16 halamanComputer Hardware SummaryFlorie Capales-PelinBelum ada peringkat
- WS 09 Bahan Ajar Function Keys ARPADokumen54 halamanWS 09 Bahan Ajar Function Keys ARPAnana kuaatBelum ada peringkat
- Computer: Commonly Operating Machine Particularly Used For Trade, Education & ResearchDokumen31 halamanComputer: Commonly Operating Machine Particularly Used For Trade, Education & ResearchAmeesh NagrathBelum ada peringkat
- v3.08 Mill Operators ManualDokumen296 halamanv3.08 Mill Operators ManualRaymond LO OtucopiBelum ada peringkat
- Sid-Meiers-Civilization Manual DOS EN PDFDokumen126 halamanSid-Meiers-Civilization Manual DOS EN PDFterranova004Belum ada peringkat
- MouseDokumen20 halamanMouseFroylan01Belum ada peringkat
- JS1 Computer Studies Examination (Third Term)Dokumen2 halamanJS1 Computer Studies Examination (Third Term)Ejiro Ndifereke100% (5)
- d2101639sg MarpossDokumen182 halamand2101639sg MarpossLeandro BomerBelum ada peringkat
- Flexi HelpDokumen189 halamanFlexi HelpJuan Martínez GonzálezBelum ada peringkat
- Second Summative Test in ComputerDokumen3 halamanSecond Summative Test in ComputerEllaine Jill CaluagBelum ada peringkat
- Keyboard Command and Special FeaturesDokumen13 halamanKeyboard Command and Special FeaturesMoch Raka RizkiBelum ada peringkat
- Manual Roland Fantom-GDokumen21 halamanManual Roland Fantom-Gsamuel BalbinoBelum ada peringkat
- Serato Sample Software ManualDokumen17 halamanSerato Sample Software ManualAnand Davis100% (1)
- Deep Sea Electronics: DSE60xx Series PC Configuration SuiteDokumen56 halamanDeep Sea Electronics: DSE60xx Series PC Configuration SuiteNelzon Naveros Loa100% (1)
- Understanding Optical Mice White PaperDokumen6 halamanUnderstanding Optical Mice White PaperSiulfer MosqueiraBelum ada peringkat
- TentaclePlayer Guide 0.9 PDFDokumen25 halamanTentaclePlayer Guide 0.9 PDFJhonatan Neptali Padron RosasBelum ada peringkat
- PMGQuadpad Manual E060308-1 (WLAN-Winbond Module)Dokumen73 halamanPMGQuadpad Manual E060308-1 (WLAN-Winbond Module)Florin TrifBelum ada peringkat
- Computer Basics For Kids:: Just How Does A Computer Work?Dokumen61 halamanComputer Basics For Kids:: Just How Does A Computer Work?Galuh PurwaBelum ada peringkat
- The Compete Ccna 200-301 Study Guide: Network Engineering EditionDari EverandThe Compete Ccna 200-301 Study Guide: Network Engineering EditionPenilaian: 5 dari 5 bintang5/5 (4)
- Hacking: A Beginners Guide To Your First Computer Hack; Learn To Crack A Wireless Network, Basic Security Penetration Made Easy and Step By Step Kali LinuxDari EverandHacking: A Beginners Guide To Your First Computer Hack; Learn To Crack A Wireless Network, Basic Security Penetration Made Easy and Step By Step Kali LinuxPenilaian: 4.5 dari 5 bintang4.5/5 (67)
- Evaluation of Some Websites that Offer Virtual Phone Numbers for SMS Reception and Websites to Obtain Virtual Debit/Credit Cards for Online Accounts VerificationsDari EverandEvaluation of Some Websites that Offer Virtual Phone Numbers for SMS Reception and Websites to Obtain Virtual Debit/Credit Cards for Online Accounts VerificationsPenilaian: 5 dari 5 bintang5/5 (1)
- Microsoft Certified Azure Fundamentals Study Guide: Exam AZ-900Dari EverandMicrosoft Certified Azure Fundamentals Study Guide: Exam AZ-900Belum ada peringkat
- AWS Certified Cloud Practitioner Study Guide: CLF-C01 ExamDari EverandAWS Certified Cloud Practitioner Study Guide: CLF-C01 ExamPenilaian: 5 dari 5 bintang5/5 (1)
- Microsoft Azure Infrastructure Services for Architects: Designing Cloud SolutionsDari EverandMicrosoft Azure Infrastructure Services for Architects: Designing Cloud SolutionsBelum ada peringkat
- Palo Alto Networks: The Ultimate Guide To Quickly Pass All The Exams And Getting Certified. Real Practice Test With Detailed Screenshots, Answers And ExplanationsDari EverandPalo Alto Networks: The Ultimate Guide To Quickly Pass All The Exams And Getting Certified. Real Practice Test With Detailed Screenshots, Answers And ExplanationsBelum ada peringkat
- Set Up Your Own IPsec VPN, OpenVPN and WireGuard Server: Build Your Own VPNDari EverandSet Up Your Own IPsec VPN, OpenVPN and WireGuard Server: Build Your Own VPNPenilaian: 5 dari 5 bintang5/5 (1)
- Cybersecurity: The Beginner's Guide: A comprehensive guide to getting started in cybersecurityDari EverandCybersecurity: The Beginner's Guide: A comprehensive guide to getting started in cybersecurityPenilaian: 5 dari 5 bintang5/5 (2)
- Computer Networking: The Complete Beginner's Guide to Learning the Basics of Network Security, Computer Architecture, Wireless Technology and Communications Systems (Including Cisco, CCENT, and CCNA)Dari EverandComputer Networking: The Complete Beginner's Guide to Learning the Basics of Network Security, Computer Architecture, Wireless Technology and Communications Systems (Including Cisco, CCENT, and CCNA)Penilaian: 4 dari 5 bintang4/5 (4)
- ITIL® 4 Create, Deliver and Support (CDS): Your companion to the ITIL 4 Managing Professional CDS certificationDari EverandITIL® 4 Create, Deliver and Support (CDS): Your companion to the ITIL 4 Managing Professional CDS certificationPenilaian: 5 dari 5 bintang5/5 (2)
- Azure Networking: Command Line Mastery From Beginner To ArchitectDari EverandAzure Networking: Command Line Mastery From Beginner To ArchitectBelum ada peringkat
- Cybersecurity: A Simple Beginner’s Guide to Cybersecurity, Computer Networks and Protecting Oneself from Hacking in the Form of Phishing, Malware, Ransomware, and Social EngineeringDari EverandCybersecurity: A Simple Beginner’s Guide to Cybersecurity, Computer Networks and Protecting Oneself from Hacking in the Form of Phishing, Malware, Ransomware, and Social EngineeringPenilaian: 5 dari 5 bintang5/5 (40)
- Terraform for Developers: Essentials of Infrastructure Automation and ProvisioningDari EverandTerraform for Developers: Essentials of Infrastructure Automation and ProvisioningBelum ada peringkat
- AWS Certified Solutions Architect Study Guide: Associate SAA-C02 ExamDari EverandAWS Certified Solutions Architect Study Guide: Associate SAA-C02 ExamBelum ada peringkat