Anda mungkin juga menyukai
- Medicine in Brief: Name the Disease in Haiku, Tanka and ArtDari EverandMedicine in Brief: Name the Disease in Haiku, Tanka and ArtPenilaian: 5 dari 5 bintang5/5 (1)
- Acute Pulmonary Edema - NEJMDokumen4 halamanAcute Pulmonary Edema - NEJMSuzika Dewi0% (1)
- Pathophysiology of Cardiogenic Pulmonary Edema - UpToDateDokumen14 halamanPathophysiology of Cardiogenic Pulmonary Edema - UpToDateStefani AtlleBelum ada peringkat
- Pathophysiology of Cardiogenic Pulmonary Edema - UpToDateDokumen9 halamanPathophysiology of Cardiogenic Pulmonary Edema - UpToDateRoberto López MataBelum ada peringkat
- Pathophysiology of Cardiogenic Pulmonary EdemaDokumen13 halamanPathophysiology of Cardiogenic Pulmonary EdemaIrina DuceacBelum ada peringkat
- Definition of Noncardiogenic Pulmonary Edemanoncardiogenic Pulmonary Edema Is IdentifiedDokumen35 halamanDefinition of Noncardiogenic Pulmonary Edemanoncardiogenic Pulmonary Edema Is IdentifiedAya EyadBelum ada peringkat
- Noncardiogenic Pulmonary EdemaDokumen19 halamanNoncardiogenic Pulmonary EdemaCláudio CalixtoBelum ada peringkat
- Pulmonary Edema - StatPearls - NCBI BookshelfDokumen7 halamanPulmonary Edema - StatPearls - NCBI BookshelfriyanasirBelum ada peringkat
- Cor PulmonaleDokumen15 halamanCor PulmonaleRizky Zulfa Afrida100% (1)
- 3487 VJXHXVDokumen13 halaman3487 VJXHXVمحمد عقيليBelum ada peringkat
- Cor PulmonaleDokumen62 halamanCor PulmonaleAlbert NixonBelum ada peringkat
- Sclerodactyly, and Telangiectasis) Syndrome Accompany-: Cor PulmonaleDokumen2 halamanSclerodactyly, and Telangiectasis) Syndrome Accompany-: Cor PulmonaledivinaBelum ada peringkat
- CorpulmonalefinalDokumen37 halamanCorpulmonalefinalRay ReyesBelum ada peringkat
- Cor Pulmonale by Prof. Dr. Rasheed Abd El Khalek M. DDokumen45 halamanCor Pulmonale by Prof. Dr. Rasheed Abd El Khalek M. DvaishnaviBelum ada peringkat
- Noncardiogenic Pulmonary EdemaDokumen12 halamanNoncardiogenic Pulmonary EdemaIrina DuceacBelum ada peringkat
- Cor Pulmonale EmedicineDokumen16 halamanCor Pulmonale EmedicineHengki Permana PutraBelum ada peringkat
- SDL 19 Cor Pulmonale - BMS16091064Dokumen6 halamanSDL 19 Cor Pulmonale - BMS16091064Jonathan YeohBelum ada peringkat
- Pathophysiology of Cardiogenic Pulmonary EdemaDokumen8 halamanPathophysiology of Cardiogenic Pulmonary EdemaLili Fiorela CRBelum ada peringkat
- Adult Respiratory Distress SyndromeDokumen5 halamanAdult Respiratory Distress SyndromeMeh AdroidBelum ada peringkat
- Acute Pulmonary Edema: NEJM December 2005 Presentation: R2 黃志宇Dokumen25 halamanAcute Pulmonary Edema: NEJM December 2005 Presentation: R2 黃志宇Rinda Putri AnggrainiBelum ada peringkat
- Non-Cardiogenic Pulmonary EdemaDokumen7 halamanNon-Cardiogenic Pulmonary EdemadarmariantoBelum ada peringkat
- Cor PulmonaleDokumen14 halamanCor PulmonaleEvangelin MelvinBelum ada peringkat
- Pulmonary HypertensionDokumen15 halamanPulmonary HypertensionChinju CyrilBelum ada peringkat
- Copd and Cor PulmonalDokumen14 halamanCopd and Cor PulmonalAldi RafaelBelum ada peringkat
- Pulmonary ThromboembolismDokumen4 halamanPulmonary ThromboembolismValerrie NgenoBelum ada peringkat
- Acute Pumonary EdemaDokumen7 halamanAcute Pumonary EdemaLucas AresBelum ada peringkat
- Uptodate Cor Pulmonale PDFDokumen13 halamanUptodate Cor Pulmonale PDFcristianamihailaBelum ada peringkat
- Cor PulmonaleDokumen27 halamanCor PulmonaleumapathisivanBelum ada peringkat
- Management of Pulmonary EdemaDokumen42 halamanManagement of Pulmonary Edemaademato4real576Belum ada peringkat
- Pathogenesis and PathophysiologyDokumen3 halamanPathogenesis and PathophysiologyEduardoFerreiraBelum ada peringkat
- Cor Pulmonale PresentationDokumen17 halamanCor Pulmonale Presentationandi reskifaisBelum ada peringkat
- Pulmonary HemorrhageDokumen6 halamanPulmonary HemorrhageSubas SharmaBelum ada peringkat
- Ali A Sovari, MD: Author:, Staff Physician, Department of Internal MedicineDokumen9 halamanAli A Sovari, MD: Author:, Staff Physician, Department of Internal MedicineMaríaDelPilarAguilarBelum ada peringkat
- Acute Pulmonary OdemaDokumen9 halamanAcute Pulmonary OdemaAnonymous ysrxggk21cBelum ada peringkat
- 1 FullDokumen4 halaman1 FullMuli YamanBelum ada peringkat
- Objectives:: Access Free Multiple Choice Questions On This Topic. Go ToDokumen6 halamanObjectives:: Access Free Multiple Choice Questions On This Topic. Go ToClarisse Cloie LamberteBelum ada peringkat
- Pulmonary Edema Facts What Causes Pulmonary Edema?Dokumen23 halamanPulmonary Edema Facts What Causes Pulmonary Edema?Louella RamosBelum ada peringkat
- Portopulmonary Hypertension and Hepatopulmonary Syndrome: ReviewDokumen8 halamanPortopulmonary Hypertension and Hepatopulmonary Syndrome: ReviewAdel HamadaBelum ada peringkat
- Kuliah 4 Oedem Paru PulmonaryedemaDokumen43 halamanKuliah 4 Oedem Paru PulmonaryedematrianaamaliaBelum ada peringkat
- Cor Pulmonale - StatPearls - NCBI BookshelfDokumen4 halamanCor Pulmonale - StatPearls - NCBI BookshelfAldi RafaelBelum ada peringkat
- Cardiac TamponadeDokumen6 halamanCardiac TamponadeVicky XieBelum ada peringkat
- Eisenmenger SyndromeDokumen9 halamanEisenmenger SyndromesyifandikaBelum ada peringkat
- Fluid Management in Pre-EclampsiaDokumen13 halamanFluid Management in Pre-Eclampsiakiki pransiskaBelum ada peringkat
- Respiratory Distress - Tintanalli's Emergency MedicineDokumen31 halamanRespiratory Distress - Tintanalli's Emergency MedicineRon KBelum ada peringkat
- Anglisht 2Dokumen8 halamanAnglisht 2Ejona SuloBelum ada peringkat
- Pulmonary Edema: René P. Michel, Peter GoldbergDokumen34 halamanPulmonary Edema: René P. Michel, Peter GoldbergNadya NovianiBelum ada peringkat
- Background: EtiologyDokumen5 halamanBackground: Etiologylia lykimBelum ada peringkat
- Pulmonary Embolism (PE) - Pulmonary Disorders - MSD Manual Professional EditionDokumen25 halamanPulmonary Embolism (PE) - Pulmonary Disorders - MSD Manual Professional Editionpeterpavel112Belum ada peringkat
- Edema ParuDokumen4 halamanEdema ParuSri BayaniBelum ada peringkat
- Cor Pulmonale ReDokumen35 halamanCor Pulmonale ReFabb NelsonBelum ada peringkat
- 3.3 Pathophysiology of Plasma LeakDokumen1 halaman3.3 Pathophysiology of Plasma LeakM Adil AliBelum ada peringkat
- Pulmonary Embolism Is A Common and Potentially Lethal ConditionDokumen13 halamanPulmonary Embolism Is A Common and Potentially Lethal ConditionJasleen KaurBelum ada peringkat
- Cardiac AsthmaDokumen12 halamanCardiac AsthmaNeupane KsabBelum ada peringkat
- Acute Respiratory Distress SyndromeDokumen36 halamanAcute Respiratory Distress Syndromedr9348345000Belum ada peringkat
- Congestive Heart FailureDokumen19 halamanCongestive Heart FailureIlavenil PanduranganBelum ada peringkat
- Bookshelf NBK360Dokumen3 halamanBookshelf NBK360Atikah PurnamasariBelum ada peringkat
- KP 2.5.5.3 Cor PulmonaleDokumen17 halamanKP 2.5.5.3 Cor Pulmonalenurul ramadhiniBelum ada peringkat
- Cardiogenic Shock: Physical FindingsDokumen9 halamanCardiogenic Shock: Physical FindingsEduardo Perez GonzalezBelum ada peringkat
- Pulmonary EdemaDokumen6 halamanPulmonary EdemaJelly BeanBelum ada peringkat
- Abordaje Nodulo PulmonarDokumen11 halamanAbordaje Nodulo PulmonarDore NanBelum ada peringkat
- SATI Ventilacion MecanicaDokumen389 halamanSATI Ventilacion MecanicaDore NanBelum ada peringkat
- 2021 - TorresDokumen28 halaman2021 - TorresCarlosBelum ada peringkat
- BTS Guidelines For The Investigation and Management of Pulmonary NodulesDokumen64 halamanBTS Guidelines For The Investigation and Management of Pulmonary NodulesHARIHARAN NARENDRANBelum ada peringkat
- DysrhythmiaDokumen30 halamanDysrhythmiaJJ JirapathBelum ada peringkat
- 9 Heart MuscleDokumen31 halaman9 Heart Muscleshoeb4uBelum ada peringkat
- 07 Blood Pressure Abnormality 1-8-2021 配布用Dokumen22 halaman07 Blood Pressure Abnormality 1-8-2021 配布用Lan NguyenBelum ada peringkat
- LWW BATES 10 Cardiovascular System Transcript FINALDokumen11 halamanLWW BATES 10 Cardiovascular System Transcript FINALShivamBelum ada peringkat
- Orbital AVMDokumen7 halamanOrbital AVMDedeh KurniasihBelum ada peringkat
- Nadi Pareeksha (Pulse Diagnosis) - An Authentic Scientific ApproachDokumen44 halamanNadi Pareeksha (Pulse Diagnosis) - An Authentic Scientific ApproachNaveen Kumar100% (1)
- Heart Failure Pelatihan Dokter KeluargaDokumen55 halamanHeart Failure Pelatihan Dokter KeluargaDelia NingrumBelum ada peringkat
- Central Venous PressureDokumen10 halamanCentral Venous PressureDenise EspinosaBelum ada peringkat
- Electrical System of The HeartDokumen4 halamanElectrical System of The HeartEldhaBelum ada peringkat
- Acute Coronary Syndrome in Clinical Practice: Firman B. LeksmonoDokumen48 halamanAcute Coronary Syndrome in Clinical Practice: Firman B. Leksmonojames klemens phieter phieBelum ada peringkat
- MidcabDokumen7 halamanMidcabNs PadiludinBelum ada peringkat
- K - 7 Atrial Flutter (IKA)Dokumen8 halamanK - 7 Atrial Flutter (IKA)thomasfelixBelum ada peringkat
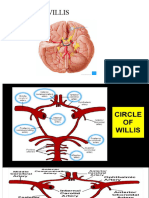
- Circle of VillisDokumen17 halamanCircle of VillisAradhana SamuelBelum ada peringkat
- Jurnal JantungDokumen5 halamanJurnal JantungFileo AdisonBelum ada peringkat
- Slide Materi Dr. Kevin Triangto, SPKFR - Heart Failure - PMR4GP 2022Dokumen27 halamanSlide Materi Dr. Kevin Triangto, SPKFR - Heart Failure - PMR4GP 2022Maulia Wisda Era ChresiaBelum ada peringkat
- Sudden Death SyndromeDokumen72 halamanSudden Death SyndromeStella CooKeyBelum ada peringkat
- Transportation in Animals and PlantsDokumen2 halamanTransportation in Animals and PlantsHimanshi JainBelum ada peringkat
- Informe DEMO Del Holter de Arritmia Contec TLC9803Dokumen14 halamanInforme DEMO Del Holter de Arritmia Contec TLC9803Edward MoralesBelum ada peringkat
- Blood and Circulation PDFDokumen47 halamanBlood and Circulation PDFeric sivaneshBelum ada peringkat
- Neuro-Ultrasonography 2020Dokumen15 halamanNeuro-Ultrasonography 2020Santiago PovedaBelum ada peringkat
- Introduction To The CvsDokumen43 halamanIntroduction To The CvsParmesh PandeyBelum ada peringkat
- Circle of WillisDokumen3 halamanCircle of WillisRisah Mae SarinoBelum ada peringkat
- Thrombosis and EmbolismDokumen39 halamanThrombosis and Embolismilva100% (1)
- Ginekologija Sa AkusertsvomDokumen2 halamanGinekologija Sa AkusertsvomZaklina RistovicBelum ada peringkat
- Angiography and ArteriographyDokumen64 halamanAngiography and ArteriographyRyBone95100% (1)
- Naskah Publikasi Agiliaaa-1Dokumen5 halamanNaskah Publikasi Agiliaaa-1Indra AlazharBelum ada peringkat
- The Heart: Test I: Read and Understand The Question Circle The Best AnswerDokumen2 halamanThe Heart: Test I: Read and Understand The Question Circle The Best AnswerHolly May MontejoBelum ada peringkat
- Medical Form Vehicle Licensing CharnwoodDokumen12 halamanMedical Form Vehicle Licensing CharnwoodKalkin43Belum ada peringkat
- Cardiovascular Agents PDFDokumen118 halamanCardiovascular Agents PDFgherlethrBelum ada peringkat
- Approach To Diagnosis of Congenital Heart DiseasesDokumen85 halamanApproach To Diagnosis of Congenital Heart DiseasesNirav CHOVATIYABelum ada peringkat