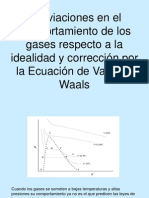
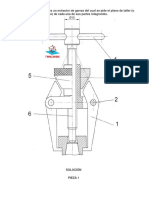
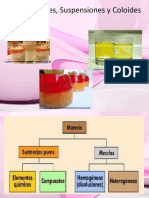
Anda mungkin juga menyukai
- Ingeniería química. Soluciones a los problemas del tomo IDari EverandIngeniería química. Soluciones a los problemas del tomo IBelum ada peringkat
- Parcial 1 ImprimirDokumen19 halamanParcial 1 ImprimirLuis F. Estrada GutierrezBelum ada peringkat
- Problemas Res. Cap 23Dokumen6 halamanProblemas Res. Cap 23Tomás HerzogBelum ada peringkat
- Sistemas Multicomponentes Grupo 02 Trabajo FinalDokumen26 halamanSistemas Multicomponentes Grupo 02 Trabajo FinalAllison JuarezBelum ada peringkat
- Termodinamica Unidad 1Dokumen12 halamanTermodinamica Unidad 1Ramm MartinezBelum ada peringkat
- Quimica General IDokumen18 halamanQuimica General ISofía Alvarez HernandezBelum ada peringkat
- Reporte de Practica Teoria Del PozoDokumen20 halamanReporte de Practica Teoria Del Pozopacopapasoterico100% (2)
- Informe de Laboratorio de Fisicoquimica 01 GasesDokumen10 halamanInforme de Laboratorio de Fisicoquimica 01 GasesJean Lopez JesusBelum ada peringkat
- Ley de Los Gases y Propiedades de Los HidrocarburosDokumen17 halamanLey de Los Gases y Propiedades de Los HidrocarburosAndrimarCorderoBelum ada peringkat
- Trabajo Final Gases Reales JMBP 180722Dokumen17 halamanTrabajo Final Gases Reales JMBP 180722jesusBelum ada peringkat
- P Sem12 Ses2 TCG-W-1-2Dokumen26 halamanP Sem12 Ses2 TCG-W-1-2Nehemias Sanchez SalasBelum ada peringkat
- Clase 1 2019-1 Gases IDEALES-REALESDokumen39 halamanClase 1 2019-1 Gases IDEALES-REALESAlberto perez william100% (1)
- Masa Molecular de Liquidos Volatiles 3Dokumen33 halamanMasa Molecular de Liquidos Volatiles 3Smith Roger Huanca Carrasco100% (2)
- Gases RealesDokumen35 halamanGases RealesMarco Cisneros,Yheferson Maza,Diego Díaz,Jairo Giohayro,David Samamé,Gino Cuadros,Ricardo Trinidad100% (1)
- Segundo Trabajo de TermodinamicaDokumen18 halamanSegundo Trabajo de TermodinamicaAlfonzo Antonio Natera OrtegaBelum ada peringkat
- Desarrollo Cuestionario de Plantas de Conversion Termica Grupo 4Dokumen60 halamanDesarrollo Cuestionario de Plantas de Conversion Termica Grupo 4Martin HerazoBelum ada peringkat
- Lab N°3 (Van Der Waals)Dokumen10 halamanLab N°3 (Van Der Waals)Luis Dani Torres GuerraBelum ada peringkat
- Ejercicios y Problemas 1 - Propiedades de Los GasesDokumen5 halamanEjercicios y Problemas 1 - Propiedades de Los GasesHeather NielsenBelum ada peringkat
- Isoterma de LangmuirDokumen13 halamanIsoterma de LangmuirAllison GarcíaBelum ada peringkat
- Taller 2 FQ TCM y Gases RespuestasDokumen7 halamanTaller 2 FQ TCM y Gases RespuestasBolaños Bolaños BolañosBelum ada peringkat
- Llaboratorio03 (1) 1Dokumen23 halamanLlaboratorio03 (1) 1DanielBelum ada peringkat
- Semana 2 FQ Gases RealesDokumen24 halamanSemana 2 FQ Gases RealesJacqueline Machón CamposBelum ada peringkat
- Informe de Gases PDFDokumen13 halamanInforme de Gases PDFCarlos Manuel Changanaqui PlasenciaBelum ada peringkat
- Monografia de Gases IdealesDokumen17 halamanMonografia de Gases IdealesSandra PerezBelum ada peringkat
- Gases Reales - ECUACION DE ESTADO - PITZERDokumen17 halamanGases Reales - ECUACION DE ESTADO - PITZERDaniel TovarBelum ada peringkat
- Problemario - Unidad - 5 Quimica AplicadaDokumen16 halamanProblemario - Unidad - 5 Quimica AplicadauzieBelum ada peringkat
- Fisicoquimica 1Dokumen29 halamanFisicoquimica 1Odalis Mallqui RiosBelum ada peringkat
- Unidad 1 Problemas ResueltosDokumen9 halamanUnidad 1 Problemas ResueltosIver Samuel Medina BBelum ada peringkat
- Masa Molecular de Liquidos Volatiles 3Dokumen33 halamanMasa Molecular de Liquidos Volatiles 3Ana Cecilia Huapaya ContrerasBelum ada peringkat
- Teoria de GasesDokumen6 halamanTeoria de GasesClaudia Mabel FloresBelum ada peringkat
- Practica #6 TermoBDokumen8 halamanPractica #6 TermoBDanielBelum ada peringkat
- Tercer Ley de La TermodinámicaDokumen54 halamanTercer Ley de La TermodinámicaDyana Alexandra SQ100% (1)
- Termo 3Dokumen36 halamanTermo 3José Emilio GuardiaBelum ada peringkat
- Informe Peso Molecular de Un Liquido VaporizableDokumen10 halamanInforme Peso Molecular de Un Liquido VaporizableAbi Abiantun DiazBelum ada peringkat
- Laboratorio Nº5Dokumen8 halamanLaboratorio Nº5CRISTHIAN TAYLOR RODRIGUEZ AYLASBelum ada peringkat
- Monografia de Fisica 1Dokumen18 halamanMonografia de Fisica 1luisBelum ada peringkat
- Trabajo Transporte - Comportamiento de GasesDokumen9 halamanTrabajo Transporte - Comportamiento de GasesRodrigo Luizaga AndiaBelum ada peringkat
- PDF Cuestionario de Plantas - CompressDokumen107 halamanPDF Cuestionario de Plantas - Compresswilson gallorBelum ada peringkat
- Fisicoquimica P2Dokumen13 halamanFisicoquimica P2Paul Silva GalvezBelum ada peringkat
- GasesDokumen6 halamanGasesJose Chavez TobarBelum ada peringkat
- Teoría de Chapman y WilkeDokumen11 halamanTeoría de Chapman y WilkeIrene Cuevas100% (2)
- Euaciones de EstadoDokumen12 halamanEuaciones de EstadoEsther Ramos GonzalezBelum ada peringkat
- Laboratorio 3 de Termodinamica OficialDokumen15 halamanLaboratorio 3 de Termodinamica OficialLuisUsseglioBelum ada peringkat
- Analis Microscopico FinalDokumen14 halamanAnalis Microscopico FinalAnonymous aEOeZkBelum ada peringkat
- Ecuación de Estado para Gases RealesDokumen13 halamanEcuación de Estado para Gases RealesCristian Ccaso MamaniBelum ada peringkat
- Relaciones Termodinamicas Parte 2Dokumen22 halamanRelaciones Termodinamicas Parte 2Maria caballeroBelum ada peringkat
- Actividad: Gases IdealesDokumen4 halamanActividad: Gases IdealesMarlenn OlánBelum ada peringkat
- Trabajo Colaborativo - Escenarios 3, 4 y 5 - SUBGRUPOS 12Dokumen14 halamanTrabajo Colaborativo - Escenarios 3, 4 y 5 - SUBGRUPOS 12derly niñoBelum ada peringkat
- Cuestionario de PlantasDokumen107 halamanCuestionario de PlantasDaniel Ruge100% (1)
- Corrección Primer Parcial de FisicoquímicaDokumen7 halamanCorrección Primer Parcial de FisicoquímicaElsie Támara ArrazolaBelum ada peringkat
- Densidad de Un GasDokumen5 halamanDensidad de Un GasTonyRamirezBelum ada peringkat
- EEVtqDokumen24 halamanEEVtqAnabel BeltránBelum ada peringkat
- Gas Real TermodinamicaDokumen6 halamanGas Real TermodinamicaSantiagoBelum ada peringkat
- Masa Molecular de Líquidos VolátilesDokumen38 halamanMasa Molecular de Líquidos VolátilesCristhianBelum ada peringkat
- Conceptos Generales de Los GasesDokumen7 halamanConceptos Generales de Los GasesJosep González100% (1)
- N° 2 Cuestionario Laboratorio Fisico QuimicaDokumen6 halamanN° 2 Cuestionario Laboratorio Fisico QuimicaRicardo CarrilloBelum ada peringkat
- Practica 3 (Actualizada)Dokumen5 halamanPractica 3 (Actualizada)basurac811Belum ada peringkat
- Teoria de Chapman y WilkeDokumen11 halamanTeoria de Chapman y WilkeAndrea NovaBelum ada peringkat
- Peso MolecularDokumen15 halamanPeso MolecularDeysiCarolinaBelum ada peringkat
- Norma IsoDokumen3 halamanNorma IsoJorge Luis Villacorta MezaBelum ada peringkat
- Pae ObstetriciaDokumen18 halamanPae ObstetriciaJorge Luis Villacorta Meza100% (2)
- Radiación DifusaDokumen7 halamanRadiación DifusaJorge Luis Villacorta MezaBelum ada peringkat
- Charla Lactancia MaternaDokumen4 halamanCharla Lactancia MaternaJorge Luis Villacorta MezaBelum ada peringkat
- EngranajesDokumen32 halamanEngranajesJorge Luis Villacorta MezaBelum ada peringkat
- El Conjunto Representado Es Un Extractor de Garras Del Cual Se Pide El Plano de TallerDokumen4 halamanEl Conjunto Representado Es Un Extractor de Garras Del Cual Se Pide El Plano de TallerJorge Luis Villacorta MezaBelum ada peringkat
- Módulo 4 Unidad 1 Trabajo ColaborativoDokumen2 halamanMódulo 4 Unidad 1 Trabajo ColaborativoJorge Luis Villacorta MezaBelum ada peringkat
- Estructura de Los Sólidos CristalinosDokumen19 halamanEstructura de Los Sólidos CristalinosJorge Luis Villacorta MezaBelum ada peringkat
- Lluvia de Ideas de Estrés en Jóvenes de 20 A 25 AñosDokumen3 halamanLluvia de Ideas de Estrés en Jóvenes de 20 A 25 AñosJorge Luis Villacorta Meza100% (1)
- Covenin 194-2019 Determinación de FibraDokumen7 halamanCovenin 194-2019 Determinación de FibraYoselyn BarreraBelum ada peringkat
- Curso de Lubricación 4100XPBDokumen34 halamanCurso de Lubricación 4100XPBJuan Carlos Rivera Navas100% (8)
- Procesos de Fabricacion 1 Desarrollo Doblado y EmbutidoDokumen9 halamanProcesos de Fabricacion 1 Desarrollo Doblado y EmbutidoFuad KhiyamiBelum ada peringkat
- 378 Conservas Vegetales MuestroDokumen7 halaman378 Conservas Vegetales Muestronmen11Belum ada peringkat
- Manejo de Semilleros de Hortalizas PDFDokumen55 halamanManejo de Semilleros de Hortalizas PDFGabriel French100% (3)
- Practica 4Dokumen14 halamanPractica 4René Acosta100% (1)
- Disoluciones y ColoidesDokumen22 halamanDisoluciones y ColoidesVicram PeraviBelum ada peringkat
- 1-Síntesis de Un Haluro de AlquiloDokumen4 halaman1-Síntesis de Un Haluro de Alquiloelbartomaster05Belum ada peringkat
- EXPLICANDO LAS TÉCNICAS DE CURADO DE LA CARNE. 10° SesionDokumen23 halamanEXPLICANDO LAS TÉCNICAS DE CURADO DE LA CARNE. 10° SesionDaniella Jimenez Moreano100% (2)
- Ajuste, Montaje, Verificación y Control de Máquina MecanismoDokumen23 halamanAjuste, Montaje, Verificación y Control de Máquina MecanismoMaritza SucaBelum ada peringkat
- Silabo de Quimica Ii - Plan 2014 PDFDokumen9 halamanSilabo de Quimica Ii - Plan 2014 PDFdugulsBelum ada peringkat
- Reconocimiento de Maquinas de Soldadura 1Dokumen7 halamanReconocimiento de Maquinas de Soldadura 1Carlos Alvarado PerezBelum ada peringkat
- Guia Campo Electrico Gauss Potencial 2016Dokumen9 halamanGuia Campo Electrico Gauss Potencial 2016Nataniel Jorquera CuevasBelum ada peringkat
- Aplicación de AzucarDokumen6 halamanAplicación de AzucarBrayan CruzBelum ada peringkat
- Práctica de Laboratorio 1Dokumen13 halamanPráctica de Laboratorio 1Leandro Quichca MedinaBelum ada peringkat
- Diseño Problemas SmithDokumen6 halamanDiseño Problemas SmithBrayan0% (1)
- Manual MicroondasDokumen32 halamanManual MicroondasEli MJ100% (1)
- (PTS) Monitoreo de Gases en MineriaDokumen6 halaman(PTS) Monitoreo de Gases en MineriaYESSICA PAULIN ALBARRACIN MORENOBelum ada peringkat
- Mineralogia en InglesDokumen8 halamanMineralogia en InglescastorcitayuniBelum ada peringkat
- Recomendaciones de Refrigerantes: Pantalla AnteriorDokumen7 halamanRecomendaciones de Refrigerantes: Pantalla AnteriorcarlosBelum ada peringkat
- Caso 4Dokumen3 halamanCaso 4Jorge OtoyaBelum ada peringkat
- Informe Química Practica 3Dokumen5 halamanInforme Química Practica 3Nicolas VallejosBelum ada peringkat
- PetroquímicoDokumen32 halamanPetroquímicoKarlos RoldanBelum ada peringkat
- Quimica IIDokumen59 halamanQuimica IIAnonymous qEAEExzNh100% (1)
- Balance de EnergiaDokumen3 halamanBalance de EnergiaAlexa SotoBelum ada peringkat
- Manual Manejo Seguro de Plaguicidas PDFDokumen10 halamanManual Manejo Seguro de Plaguicidas PDFjose luisBelum ada peringkat
- PrecipitadosDokumen31 halamanPrecipitadosMaria Rene TorresBelum ada peringkat
- Ejercicio de Aplicación DISEÑO DE UNA BOCATOMA DE FONDO.Dokumen14 halamanEjercicio de Aplicación DISEÑO DE UNA BOCATOMA DE FONDO.DavidBallestaPalaciosBelum ada peringkat
- Ensayos de Materiales PDFDokumen19 halamanEnsayos de Materiales PDFJose Francisco Agulló Mateu100% (1)
- Quiz 50Dokumen10 halamanQuiz 50RUBEN FUENTESBelum ada peringkat