Anda mungkin juga menyukai
- Checklist of Mandatory Documentation Required by ISO 13485:2016Dokumen15 halamanChecklist of Mandatory Documentation Required by ISO 13485:2016LiliBelum ada peringkat
- 18 Composition Rules For Photos That ShineDokumen20 halaman18 Composition Rules For Photos That Shinemahfuzkhan100% (1)
- ISO 13485-2016 Implementation and Compliance With MyEasyISO - R01 - 01062017Dokumen5 halamanISO 13485-2016 Implementation and Compliance With MyEasyISO - R01 - 01062017kaushal_sutariaBelum ada peringkat
- Overview of ISO 13485-2016 3rd EditionDokumen23 halamanOverview of ISO 13485-2016 3rd EditionumarBelum ada peringkat
- ISO 13485 2016 Documentation Manual Clause Wise RequirementsDokumen15 halamanISO 13485 2016 Documentation Manual Clause Wise Requirementsqmicertification100% (1)
- 227 20 Unique Device Indentifier UDI For Medical Devices Used in The Gas IndustryDokumen16 halaman227 20 Unique Device Indentifier UDI For Medical Devices Used in The Gas IndustryMauro CostaBelum ada peringkat
- ISO 13485 2003 Audit ChecklistDokumen38 halamanISO 13485 2003 Audit Checklisttousifaslam100% (1)
- Technical FilesDokumen15 halamanTechnical Fileshitham shehataBelum ada peringkat
- Design Controls For Medical Device CompaniesDokumen9 halamanDesign Controls For Medical Device CompaniesmahendranBelum ada peringkat
- Audit Checklist For ISO 13485Dokumen6 halamanAudit Checklist For ISO 13485EdBelum ada peringkat
- 2016 Ia Sample ChecklistDokumen5 halaman2016 Ia Sample ChecklistMiss BreedloveBelum ada peringkat
- ISO 13485 Internal Audit ChecklistDokumen23 halamanISO 13485 Internal Audit ChecklistGary Jheferson Salazar Rondon60% (5)
- ISO 13485 Operational Procedure QOP-73-02 (A) Design Risk ManagementDokumen4 halamanISO 13485 Operational Procedure QOP-73-02 (A) Design Risk Managementtahir_rizvi1569Belum ada peringkat
- Medical Device Iso 13485:2016Dokumen88 halamanMedical Device Iso 13485:2016Aymen Bekir75% (8)
- Mecha World Compendium Playbooks BWDokumen12 halamanMecha World Compendium Playbooks BWRobson Alves MacielBelum ada peringkat
- Bsi MD Risk Management For Medical Devices Webinar 131119 Uk enDokumen29 halamanBsi MD Risk Management For Medical Devices Webinar 131119 Uk enrakesh marwahBelum ada peringkat
- 2016 StandardDokumen39 halaman2016 StandardAnkur Dhir100% (4)
- HUMSS - Introduction To World Religions & Belief Systems CGDokumen13 halamanHUMSS - Introduction To World Religions & Belief Systems CGAliuqus SirJasper89% (18)
- Audit Checklist - IsO 13485 and MDDDokumen6 halamanAudit Checklist - IsO 13485 and MDDMarcos Poffo100% (2)
- Medical Device Quality Management Systems: Strategy and Techniques for Improving Efficiency and EffectivenessDari EverandMedical Device Quality Management Systems: Strategy and Techniques for Improving Efficiency and EffectivenessBelum ada peringkat
- Quality Manual ExampleDokumen32 halamanQuality Manual ExamplesaiaviBelum ada peringkat
- Syringe Tech. File ChecklistDokumen3 halamanSyringe Tech. File ChecklistdearistyaBelum ada peringkat
- How To Simplify Compliance With The New ISO 13485 2016 Final PDFDokumen63 halamanHow To Simplify Compliance With The New ISO 13485 2016 Final PDFAymen Bekir100% (3)
- Checklist of Mandatory Documentation Required by Iso 13485 2016 enDokumen18 halamanChecklist of Mandatory Documentation Required by Iso 13485 2016 enjmittal75% (4)
- 410 09e Checkliste For Assessment 13485 MDDDokumen51 halaman410 09e Checkliste For Assessment 13485 MDDeko Setyawan100% (1)
- ISO 13485 Documentation RequirementsDokumen2 halamanISO 13485 Documentation Requirementsalexrferreira67% (3)
- ISO 13485 Operational Procedure QOP-42-01 (A) Control of Documents PDFDokumen9 halamanISO 13485 Operational Procedure QOP-42-01 (A) Control of Documents PDFTonNuBaoNgoc100% (1)
- PMS SopDokumen8 halamanPMS Sopstevekent40% (5)
- Analysis of Vitamins A and E by HPLCDokumen9 halamanAnalysis of Vitamins A and E by HPLCamit545Belum ada peringkat
- Inspección, Pruebas, Y Mantenimiento de Gabinetes de Ataque Rápido E HidrantesDokumen3 halamanInspección, Pruebas, Y Mantenimiento de Gabinetes de Ataque Rápido E HidrantesVICTOR RALPH FLORES GUILLENBelum ada peringkat
- Iso 13485 & 21 CFR 820 Template Documentation Operational Procedure Qop 42 01 Control of DocumentsDokumen9 halamanIso 13485 & 21 CFR 820 Template Documentation Operational Procedure Qop 42 01 Control of DocumentsfattuatiBelum ada peringkat
- ISO 13485 Audit ChecklistDokumen38 halamanISO 13485 Audit ChecklistRRR1Belum ada peringkat
- Pecb Iso 13485 Lead Auditor Exam Preparation GuideDokumen15 halamanPecb Iso 13485 Lead Auditor Exam Preparation GuiderberrospiBelum ada peringkat
- Tuma Research ManualDokumen57 halamanTuma Research ManualKashinde Learner Centered Mandari100% (1)
- Gap Analysis ISO 13485 2016 - ISO 9001 2015Dokumen36 halamanGap Analysis ISO 13485 2016 - ISO 9001 2015Pooja Sankhla80% (5)
- Checkliste en Iso 13485-2016 eDokumen2 halamanCheckliste en Iso 13485-2016 eKlaudija LutovskaBelum ada peringkat
- Device Master RecordDokumen1 halamanDevice Master RecordkuttyjBelum ada peringkat
- AbstractDokumen11 halamanAbstractMaurizio FieraBelum ada peringkat
- QP-007 Risk Management ProcessDokumen12 halamanQP-007 Risk Management Processesraa asemBelum ada peringkat
- Quality Control of Rigid Pavements 1Dokumen58 halamanQuality Control of Rigid Pavements 1pranjpatil100% (1)
- ISO 13485 Version 2016 Requirements NotesDokumen24 halamanISO 13485 Version 2016 Requirements Notesda_reaper_dasBelum ada peringkat
- MDR Initial Certification Checklist (Found On Elsmar)Dokumen25 halamanMDR Initial Certification Checklist (Found On Elsmar)quality4720 GOLNIT50% (2)
- Quality Management System Manual: QM-00 Index (Full Text)Dokumen5 halamanQuality Management System Manual: QM-00 Index (Full Text)Quality and Safety Consultants Co.Belum ada peringkat
- NAV SOLVING PROBLEM 3 (1-20) .PpsDokumen37 halamanNAV SOLVING PROBLEM 3 (1-20) .Ppsmsk5in100% (1)
- ISO 13485 Quality Management System A Complete Guide - 2020 EditionDari EverandISO 13485 Quality Management System A Complete Guide - 2020 EditionBelum ada peringkat
- Iso 13485 Manual DocumentsDokumen14 halamanIso 13485 Manual DocumentsRRR1100% (1)
- ISO 13485 UpdateDokumen43 halamanISO 13485 Updatehitham shehata100% (1)
- Iso 9001 Iso 13485 QMS MatrixDokumen1 halamanIso 9001 Iso 13485 QMS MatrixmhkeerthiBelum ada peringkat
- FDA Medical Device LabelingDokumen54 halamanFDA Medical Device LabelingYachao LiuBelum ada peringkat
- Medical Device Risk Management ISO 14971: Dan O'Leary President Ombu Enterprises, LLCDokumen86 halamanMedical Device Risk Management ISO 14971: Dan O'Leary President Ombu Enterprises, LLC7lightbourn5893Belum ada peringkat
- MDR Readiness Checklist: Prepared by Cite Medical SolutionsDokumen37 halamanMDR Readiness Checklist: Prepared by Cite Medical SolutionsBeal100% (1)
- Iso13485 QOP4101 RiskMng PDFDokumen4 halamanIso13485 QOP4101 RiskMng PDFQuality and Safety Consultants Co.Belum ada peringkat
- ISO 13485 2016 - Vs - FDA 21 CRF Part 820Dokumen6 halamanISO 13485 2016 - Vs - FDA 21 CRF Part 820nasa4sunBelum ada peringkat
- Institutional Group Agencies For EducationDokumen22 halamanInstitutional Group Agencies For EducationGlory Aroma100% (1)
- ISO 13485 2003 Vs ISO 13485 2016 Matrix ENDokumen13 halamanISO 13485 2003 Vs ISO 13485 2016 Matrix ENHilario Alinabon100% (1)
- ISO 13485 Process MatrixDokumen2 halamanISO 13485 Process MatrixManjunath BBelum ada peringkat
- Usability Best PracticesDokumen37 halamanUsability Best PracticesMashal PkBelum ada peringkat
- QP19-Vigilance Report - CE MarkDokumen18 halamanQP19-Vigilance Report - CE Markanusha shankarBelum ada peringkat
- ISO 13485 Process Matrix ExampleDokumen3 halamanISO 13485 Process Matrix Examplesaiavi100% (1)
- Alchemy of The HeartDokumen7 halamanAlchemy of The HeartAbdul RahimBelum ada peringkat
- Contoh Exposition TextDokumen1 halamanContoh Exposition TextKristin SeranBelum ada peringkat
- Standard Awareness Training 13485Dokumen21 halamanStandard Awareness Training 13485Jeelani BashaBelum ada peringkat
- EU MDR - Pinnacle Software Technologies LimitedDokumen6 halamanEU MDR - Pinnacle Software Technologies LimitedPinnacle Software Technologies Limited100% (1)
- 13485-2016 Numbering Gap Analysis With Comments - ElsmarDokumen3 halaman13485-2016 Numbering Gap Analysis With Comments - ElsmarBharathBelum ada peringkat
- Mandatory Documents and Records Required by ISO 13485Dokumen7 halamanMandatory Documents and Records Required by ISO 13485Ricky MarkBelum ada peringkat
- Meddev 2.5-10 Guideline For Authorised Representatives January 2012Dokumen17 halamanMeddev 2.5-10 Guideline For Authorised Representatives January 2012amit545Belum ada peringkat
- Import of Transgenic Planting MaterialDokumen5 halamanImport of Transgenic Planting Materialamit545Belum ada peringkat
- PolymorhismDokumen3 halamanPolymorhismamit545Belum ada peringkat
- ENVIRONMENTAL RESOURCE1 The Allocative Characteristics of Environmental ResourcesDokumen11 halamanENVIRONMENTAL RESOURCE1 The Allocative Characteristics of Environmental Resourcesamit545Belum ada peringkat
- SDS PageDokumen5 halamanSDS Pageamit545Belum ada peringkat
- Endangered Medicinal Plant Species in Himachal PradeshDokumen2 halamanEndangered Medicinal Plant Species in Himachal Pradeshamit545Belum ada peringkat
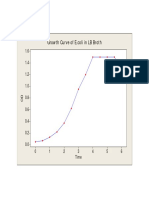
- Growth Curve of E.coliDokumen1 halamanGrowth Curve of E.coliamit545Belum ada peringkat
- Experimental Analysis FunctionalDokumen4 halamanExperimental Analysis Functionalamit545Belum ada peringkat
- Environmental Resource5 Environmental Impact AssesmentsDokumen6 halamanEnvironmental Resource5 Environmental Impact Assesmentsamit545Belum ada peringkat
- Sesquiterpene Lactones From Elephantopus ScaberDokumen1 halamanSesquiterpene Lactones From Elephantopus Scaberamit545Belum ada peringkat
- Page 1 of 1 EIA (Environmental Impact Assessment) Practice in IndiaDokumen1 halamanPage 1 of 1 EIA (Environmental Impact Assessment) Practice in Indiaamit545Belum ada peringkat
- Correspondence Between ISO 13485Dokumen2 halamanCorrespondence Between ISO 13485amit545Belum ada peringkat
- HCCG ES10 - Suture Removal Specification 2015Dokumen4 halamanHCCG ES10 - Suture Removal Specification 2015amit545Belum ada peringkat
- Arginine in DiseaseDokumen2 halamanArginine in Diseaseamit545Belum ada peringkat
- Knowledge Gap in DiabetesDokumen3 halamanKnowledge Gap in Diabetesamit545Belum ada peringkat
- Correspondence Between ISO 13485Dokumen2 halamanCorrespondence Between ISO 13485amit545Belum ada peringkat
- Word CountDokumen3 halamanWord CountLeo LonardelliBelum ada peringkat
- Yale Revision WorksheetDokumen3 halamanYale Revision WorksheetYASHI AGRAWALBelum ada peringkat
- Dating Apps MDokumen2 halamanDating Apps Mtuanhmt040604Belum ada peringkat
- Toolbox TalkDokumen14 halamanToolbox Talkcall_mustafas2361Belum ada peringkat
- Session 1Dokumen18 halamanSession 1Akash GuptaBelum ada peringkat
- Iguard® LM SeriesDokumen82 halamanIguard® LM SeriesImran ShahidBelum ada peringkat
- Quotation of Suny PDFDokumen5 halamanQuotation of Suny PDFHaider KingBelum ada peringkat
- IR2153 Parte6Dokumen1 halamanIR2153 Parte6FRANK NIELE DE OLIVEIRABelum ada peringkat
- SW OSDokumen11 halamanSW OSErnest OfosuBelum ada peringkat
- CATaclysm Preview ReleaseDokumen52 halamanCATaclysm Preview ReleaseGhaderalBelum ada peringkat
- 4.2.4.5 Packet Tracer - Connecting A Wired and Wireless LAN InstructionsDokumen5 halaman4.2.4.5 Packet Tracer - Connecting A Wired and Wireless LAN InstructionsAhmadHijaziBelum ada peringkat
- Isi Rumen SBG Subtitusi HijauanDokumen3 halamanIsi Rumen SBG Subtitusi HijauanBagas ImamsyahBelum ada peringkat
- Immunity Question Paper For A Level BiologyDokumen2 halamanImmunity Question Paper For A Level BiologyJansi Angel100% (1)
- Chapter 13 CarbohydratesDokumen15 halamanChapter 13 CarbohydratesShanna Sophia PelicanoBelum ada peringkat
- State Space ModelsDokumen19 halamanState Space Modelswat2013rahulBelum ada peringkat
- ReadingDokumen205 halamanReadingHiền ThuBelum ada peringkat
- Is 2 - 2000 Rules For Rounded Off For Numericals PDFDokumen18 halamanIs 2 - 2000 Rules For Rounded Off For Numericals PDFbala subramanyamBelum ada peringkat
- Practice Problems - Electrochemical CellDokumen5 halamanPractice Problems - Electrochemical CellYehia IbrahimBelum ada peringkat
- DB Lecture Note All in ONEDokumen85 halamanDB Lecture Note All in ONEyonasante2121Belum ada peringkat
- Borges, The SouthDokumen4 halamanBorges, The Southdanielg233100% (1)