Anda mungkin juga menyukai
- Manual técnico de refrigerantesDari EverandManual técnico de refrigerantesPenilaian: 4 dari 5 bintang4/5 (4)
- Examen y Respuesta de ISO 9001-2015. V1.0Dokumen7 halamanExamen y Respuesta de ISO 9001-2015. V1.0Ruben Perez Ayo80% (5)
- Viscosidad de fluidos: tabla de conversión cps, Poise, cStDokumen1 halamanViscosidad de fluidos: tabla de conversión cps, Poise, cStGenesisQuiroz100% (2)
- Viscosidad de fluidos: tabla de conversión cps, Poise, cStDokumen1 halamanViscosidad de fluidos: tabla de conversión cps, Poise, cStGenesisQuiroz100% (2)
- Viscosidad de fluidos: tabla de conversión cps, Poise, cStDokumen1 halamanViscosidad de fluidos: tabla de conversión cps, Poise, cStGenesisQuiroz100% (2)
- ASTM D 1945 en EspañolDokumen37 halamanASTM D 1945 en EspañolRuben Perez Ayo100% (3)
- Flujo de fluidos e intercambio de calorDari EverandFlujo de fluidos e intercambio de calorPenilaian: 1 dari 5 bintang1/5 (1)
- Nom 026 StpsDokumen46 halamanNom 026 StpsDaniel Hernandez Parra100% (1)
- Tabla Astm 5bDokumen5 halamanTabla Astm 5bRuben Perez AyoBelum ada peringkat
- Norma D - 4530Dokumen11 halamanNorma D - 4530Luis Ernesto Marin JaimesBelum ada peringkat
- Estudio Del Tercer Paso PDFDokumen2 halamanEstudio Del Tercer Paso PDFJesus Octavio Garcia Saavedra100% (1)
- La Integridad CristianaDokumen14 halamanLa Integridad CristianaNoemi De Gil100% (1)
- Astm D 2710-92 Método de Ensayo Estándar para El Indice de Bromo de Hidrocarburos de Petroleo Por Valoración Electro-MetricaDokumen5 halamanAstm D 2710-92 Método de Ensayo Estándar para El Indice de Bromo de Hidrocarburos de Petroleo Por Valoración Electro-MetricaRuben Perez AyoBelum ada peringkat
- Destilación Atmosférica y Al VacíoDokumen29 halamanDestilación Atmosférica y Al VacíoHectorRColombiaBelum ada peringkat
- Recuperacion de Azufre Informe Final Final Jajaja-1 - ColmenaDokumen11 halamanRecuperacion de Azufre Informe Final Final Jajaja-1 - ColmenaAna ClaudiaBelum ada peringkat
- Grua Giratoria 1Dokumen34 halamanGrua Giratoria 1garritakidd100% (3)
- Lista de Chequeo NC ISO - IEC 17025 - 2005 PDFDokumen6 halamanLista de Chequeo NC ISO - IEC 17025 - 2005 PDFRuben Perez AyoBelum ada peringkat
- Lista de Chequeo NC ISO - IEC 17025 - 2005 PDFDokumen6 halamanLista de Chequeo NC ISO - IEC 17025 - 2005 PDFRuben Perez AyoBelum ada peringkat
- Titulaciones Volumetricas de NeutralizacionDokumen13 halamanTitulaciones Volumetricas de NeutralizacionPaucar Coyla JonathanBelum ada peringkat
- Astm D 2784 Azufre en GLPDokumen13 halamanAstm D 2784 Azufre en GLPRuben Perez AyoBelum ada peringkat
- Ecuaciones de estado para gases ideales y realesDokumen42 halamanEcuaciones de estado para gases ideales y realesmaximiliano_molin_21Belum ada peringkat
- Estimacion de Propiedades Del Petroleo y Sus Productos Vol. 1Dokumen227 halamanEstimacion de Propiedades Del Petroleo y Sus Productos Vol. 1juan pablo diaz loeza100% (1)
- Significado de Los KatasDokumen6 halamanSignificado de Los KatasFelipe GutierrezBelum ada peringkat
- Eco DomoDokumen5 halamanEco DomoNi Neu AbizenaBelum ada peringkat
- Prueba de Salinidad en El CrudoDokumen12 halamanPrueba de Salinidad en El CrudoDavid GPBelum ada peringkat
- Sistemas de Desalado de Crudo 3Dokumen17 halamanSistemas de Desalado de Crudo 3wilmar100% (1)
- ANTONIO CORNEJO POLAR - Mestizaje, Transculturación, HeterogeneidadDokumen2 halamanANTONIO CORNEJO POLAR - Mestizaje, Transculturación, HeterogeneidadEvillyn RodriguesBelum ada peringkat
- EXAMEN - Interpretacion de La Norma ISO 17025-2006Dokumen7 halamanEXAMEN - Interpretacion de La Norma ISO 17025-2006Ruben Perez Ayo100% (1)
- Cancionero VillancicosDokumen8 halamanCancionero VillancicosMiguel Leandro100% (2)
- M1-5b SeparadoresDokumen117 halamanM1-5b Separadoresalpalo87100% (1)
- Método de Prueba Estándar para Azufre en Productos de PetróleoDokumen21 halamanMétodo de Prueba Estándar para Azufre en Productos de PetróleoMalu AisneBelum ada peringkat
- NMX L 145 Scfi 2004 Secuestrante de h2sDokumen15 halamanNMX L 145 Scfi 2004 Secuestrante de h2sEnrique Alejandro Ovando100% (1)
- Cloruros OrganicosDokumen16 halamanCloruros OrganicosRoberto Eduardo Salvador MonteroBelum ada peringkat
- Clasificación de Los Hidrocarburos y Determinación Del Contenido de AzufreDokumen65 halamanClasificación de Los Hidrocarburos y Determinación Del Contenido de AzufreYullyMayerlyRincónMartínez100% (1)
- Ecuaciones de EstadoDokumen7 halamanEcuaciones de EstadoKendy HernandezBelum ada peringkat
- Aplicación Exitosa de La Inyección Cíclica de Gas Campo ProvinciaDokumen13 halamanAplicación Exitosa de La Inyección Cíclica de Gas Campo ProvinciaEdgar GonzalezBelum ada peringkat
- Astm D 1298 Api PDFDokumen7 halamanAstm D 1298 Api PDFSALOMONBelum ada peringkat
- Analisis Del PetroleoDokumen2 halamanAnalisis Del Petroleoyair vargasBelum ada peringkat
- Consulta Normas de ViscosidadDokumen7 halamanConsulta Normas de ViscosidadStalin Chugchilán100% (1)
- ASTM - A249 - Prueba de Corrosión IntergranularDokumen2 halamanASTM - A249 - Prueba de Corrosión IntergranularalexgrraBelum ada peringkat
- EDUCACION FET Actualizacion Tecnologica 3Dokumen29 halamanEDUCACION FET Actualizacion Tecnologica 3Cristian Diego Cruz TejerinaBelum ada peringkat
- Astm D-2622 (Determinación de Azufre Por Fluorescencia de Rayos Xtwin-X)Dokumen20 halamanAstm D-2622 (Determinación de Azufre Por Fluorescencia de Rayos Xtwin-X)JOSEBelum ada peringkat
- Determinación de Acidez Total de Acido Sulfurico Industrial Segun Astm E223-08Dokumen8 halamanDeterminación de Acidez Total de Acido Sulfurico Industrial Segun Astm E223-08Wilfredo Jhon Arteaga HualiBelum ada peringkat
- Propiedades Del Gas NaturalDokumen34 halamanPropiedades Del Gas NaturalRafael SanguezaBelum ada peringkat
- Laboratorio para Medicion de La ViscosidadDokumen18 halamanLaboratorio para Medicion de La ViscosidadSergioAndresRamonBelum ada peringkat
- Contenido de Azufre (Lab Fluidos)Dokumen35 halamanContenido de Azufre (Lab Fluidos)Heberth Julian RodriguezBelum ada peringkat
- Marco TeoricoDokumen2 halamanMarco TeoricoJunior Barcianovich DelgadovBelum ada peringkat
- Determinación de la densidad de la gasolina mediante el método del picnómetroDokumen17 halamanDeterminación de la densidad de la gasolina mediante el método del picnómetroElmer Guevara VasquezBelum ada peringkat
- Rubiales (Viscosidad)Dokumen15 halamanRubiales (Viscosidad)Punto YanbalBelum ada peringkat
- Algoritmo AGA 7 para medición de flujo de gas naturalDokumen12 halamanAlgoritmo AGA 7 para medición de flujo de gas naturalDiego Fabian Ruiz VacaflorBelum ada peringkat
- Flash and Fire PointDokumen25 halamanFlash and Fire PointWilliam ContrerasBelum ada peringkat
- Gas Properties-Grupo 3Dokumen13 halamanGas Properties-Grupo 3Edilson Montilla100% (1)
- Prueba de Eficiencia Homogenizadores Jgr-ccs-TosDokumen6 halamanPrueba de Eficiencia Homogenizadores Jgr-ccs-TosAnonymous i0VwbnaOWBelum ada peringkat
- Aleaciones Especiales AcabadoDokumen10 halamanAleaciones Especiales AcabadoErick Montenegro100% (1)
- Analisis SARADokumen2 halamanAnalisis SARADaniel Buitrón0% (1)
- PTE - Metales ASTM 5863Dokumen10 halamanPTE - Metales ASTM 5863Luis Ernesto Marin JaimesBelum ada peringkat
- Presentacion de Vapor de ReidDokumen10 halamanPresentacion de Vapor de Reidjjdv3755Belum ada peringkat
- Informe de SalinidadDokumen15 halamanInforme de SalinidadPaula Alvarez MolinaBelum ada peringkat
- Deshidratación de gas natural con desecantesDokumen8 halamanDeshidratación de gas natural con desecantesRuben Alfred HuantoBelum ada peringkat
- Remocion de Mercurio Del Gas Natural - En.esDokumen1 halamanRemocion de Mercurio Del Gas Natural - En.esMarcelo BlasBelum ada peringkat
- nmx-k-073-1987 AcidezDokumen6 halamannmx-k-073-1987 AcidezarturoBelum ada peringkat
- 2 GPK-PRM-02 Medicion de Temperatura (Termometro Electronico)Dokumen3 halaman2 GPK-PRM-02 Medicion de Temperatura (Termometro Electronico)Julian Felipe Noguera CruzBelum ada peringkat
- Tratamiento de Gases Usando MDEA Versio 97-2003Dokumen28 halamanTratamiento de Gases Usando MDEA Versio 97-2003zabalamgBelum ada peringkat
- Tanques Refrigerados TeoriaDokumen2 halamanTanques Refrigerados TeoriaSof RomeroBelum ada peringkat
- Npr3098 - Manual Sos Mmx219dc225p A Fl150 Tq300 NT Si Gab Fe - Rev 0Dokumen57 halamanNpr3098 - Manual Sos Mmx219dc225p A Fl150 Tq300 NT Si Gab Fe - Rev 0Danitza Ivanna MendezBelum ada peringkat
- Análisis de Humedad Y Cenizas en AlimentosDokumen2 halamanAnálisis de Humedad Y Cenizas en AlimentosElvinDaniloPincayCataguaBelum ada peringkat
- 73534-8464-Proyecto TransversalDokumen11 halaman73534-8464-Proyecto TransversalJenniBelum ada peringkat
- Comportamiento de Sistemas GaseososDokumen5 halamanComportamiento de Sistemas GaseososManuelAndresParraMuñoz100% (4)
- Calor LatenteDokumen13 halamanCalor LatenteJosé TorrezBelum ada peringkat
- 2 Captacion, Separacion y Compresion 2009Dokumen66 halaman2 Captacion, Separacion y Compresion 2009David Knutti VidaurreBelum ada peringkat
- Caracterizacion CRUDO BoliviaDokumen2 halamanCaracterizacion CRUDO BoliviaDenis Gonzales Vasquez100% (1)
- Diseño de Redes de Gas PDFDokumen397 halamanDiseño de Redes de Gas PDFarturoBelum ada peringkat
- Valoracion Tecnologica Del PetroleoDokumen43 halamanValoracion Tecnologica Del PetroleoVanessa M.Belum ada peringkat
- Hidratos de Metano INFORMEDokumen36 halamanHidratos de Metano INFORMEJonathan Fernando Lizana Figueroa33% (3)
- Asignacion 1 Acido OxalicoDokumen51 halamanAsignacion 1 Acido OxalicoCarolina Botero RodriguezBelum ada peringkat
- Seguridad en RefineríasDokumen3 halamanSeguridad en RefineríasJ Daniel PereyraBelum ada peringkat
- Proyecto de TesisDokumen15 halamanProyecto de TesisMarcoDulongJaraBelum ada peringkat
- ASTM D 1548-92 Método de Prueba Patrón para Vanadio en Petróleo Combustible PesadoDokumen7 halamanASTM D 1548-92 Método de Prueba Patrón para Vanadio en Petróleo Combustible PesadoRuben Perez Ayo100% (1)
- Estudios de Precision R y RDokumen5 halamanEstudios de Precision R y RRuben Perez AyoBelum ada peringkat
- Procesos de RefinacionDokumen24 halamanProcesos de RefinacionBrandon Gonzalez100% (1)
- Determinación de Aceites y Grasas en Agua PDFDokumen2 halamanDeterminación de Aceites y Grasas en Agua PDFRuben Perez AyoBelum ada peringkat
- ISO/IEC 17025:2017 requisitos laboratorios ensayos calibraciónDokumen1 halamanISO/IEC 17025:2017 requisitos laboratorios ensayos calibraciónRuben Perez Ayo100% (1)
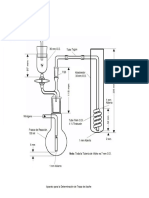
- Aparato Deter Traza de AzufreDokumen1 halamanAparato Deter Traza de AzufreRuben Perez AyoBelum ada peringkat
- Ejemplo-Agregar Min y Max A Gráfico en ExcelDokumen1 halamanEjemplo-Agregar Min y Max A Gráfico en ExcelRuben Perez AyoBelum ada peringkat
- Determinación de Aceites y Grasas en Agua PDFDokumen2 halamanDeterminación de Aceites y Grasas en Agua PDFRuben Perez AyoBelum ada peringkat
- Ejemplo-Agregar Min y Max A Gráfico en ExcelDokumen1 halamanEjemplo-Agregar Min y Max A Gráfico en ExcelRuben Perez AyoBelum ada peringkat
- Determinacion de Contaminacion Por Particulas Solidas en Combustibel de AviacionDokumen9 halamanDeterminacion de Contaminacion Por Particulas Solidas en Combustibel de AviacionRuben Perez Ayo100% (2)
- Calcular - % Evaporado A Temp PrescritaDokumen4 halamanCalcular - % Evaporado A Temp PrescritaRuben Perez Ayo100% (1)
- Tutwiler ApparatusDokumen1 halamanTutwiler ApparatusRuben Perez AyoBelum ada peringkat
- Sulfuro de Hidrogeno en Gases Por El Metodo TutwilerDokumen4 halamanSulfuro de Hidrogeno en Gases Por El Metodo TutwilerRuben Perez Ayo100% (2)
- Estudios de Precision R y RDokumen5 halamanEstudios de Precision R y RRuben Perez AyoBelum ada peringkat
- Análisis de Dureza Total Por Titulación Con EDTADokumen5 halamanAnálisis de Dureza Total Por Titulación Con EDTARuben Perez AyoBelum ada peringkat
- Norma VOP 357-64 Trazas de AzufreDokumen6 halamanNorma VOP 357-64 Trazas de AzufreRuben Perez AyoBelum ada peringkat
- Como Reducir El Tamano de Un Archivo PDF 29193 Ok23tqDokumen2 halamanComo Reducir El Tamano de Un Archivo PDF 29193 Ok23tqRuben Perez AyoBelum ada peringkat
- Quemador Mark 7 para EAA SpetrAA220Dokumen1 halamanQuemador Mark 7 para EAA SpetrAA220Ruben Perez AyoBelum ada peringkat
- Nebulizador Varian SpectraAA220Dokumen3 halamanNebulizador Varian SpectraAA220Ruben Perez AyoBelum ada peringkat
- Sedimento Tota lIS0 10307-2 PDFDokumen4 halamanSedimento Tota lIS0 10307-2 PDFRuben Perez AyoBelum ada peringkat
- A Busca Do Ator e EspectadorDokumen79 halamanA Busca Do Ator e EspectadorVictor Paulo de SeixasBelum ada peringkat
- Diseño geométrico de vía plancha C-69Dokumen10 halamanDiseño geométrico de vía plancha C-69Camila Martínez OsmaBelum ada peringkat
- Nudos seguros para colgar hamacas y sillasDokumen2 halamanNudos seguros para colgar hamacas y sillasCarlos Contreras PeraltaBelum ada peringkat
- Mitos Cortos de LatinoaméricaDokumen14 halamanMitos Cortos de LatinoaméricaJulio Winfred Gil Campuzano100% (1)
- Detección de identidadesDokumen16 halamanDetección de identidadeslavidaexageradaBelum ada peringkat
- Explicacin Libros Colaborativos - Un Da en La PlayaDokumen4 halamanExplicacin Libros Colaborativos - Un Da en La Playaapi-266976944Belum ada peringkat
- 2 Examen Computación Uni Ges - SábadoDokumen2 halaman2 Examen Computación Uni Ges - SábadoMartin P BaylonBelum ada peringkat
- Versos Bíblicos Que Los Demonios Odian EscucharDokumen18 halamanVersos Bíblicos Que Los Demonios Odian Escucharolmecas2016Belum ada peringkat
- Escritos Del Antiguo Testamento Versus Enseñanzas de AmenemopeDokumen2 halamanEscritos Del Antiguo Testamento Versus Enseñanzas de AmenemopeenhacoreBelum ada peringkat
- Marisol PDFDokumen7 halamanMarisol PDFMireya Represa PérezBelum ada peringkat
- 1 - CrestomatiaDokumen9 halaman1 - CrestomatiaTeologos UTACBelum ada peringkat
- Vicente Fernandez GomezDokumen2 halamanVicente Fernandez Gomezmaria paula francoBelum ada peringkat
- Guía estimulación lenguaje 3-6 añosDokumen20 halamanGuía estimulación lenguaje 3-6 añosMileysi RiveraBelum ada peringkat
- Amor y Muerte en Florencia - SarahDokumen267 halamanAmor y Muerte en Florencia - SarahVallolet Mas100% (2)
- Concepto de OpenofficeDokumen6 halamanConcepto de OpenofficeNaty D' AnggelBelum ada peringkat
- El Renacimiento: Humanismo y ciudad idealDokumen2 halamanEl Renacimiento: Humanismo y ciudad idealAnonymous xDf8uEiXBelum ada peringkat
- 01ASELF Temario AyA (Nivel 1) SEIS OviedoDokumen38 halaman01ASELF Temario AyA (Nivel 1) SEIS OviedoNachoVillaverdeBelum ada peringkat
- 6°segundo Cuadernillo ProvinciaDokumen44 halaman6°segundo Cuadernillo ProvinciaAle LemonnierBelum ada peringkat
- Propiedades de Los ElementosDokumen20 halamanPropiedades de Los ElementosLuis AranoBelum ada peringkat
- Versos Descontrolados TinkuyDokumen11 halamanVersos Descontrolados TinkuyfavillescoBelum ada peringkat
- Amiga Mia Michel El BuenonDokumen2 halamanAmiga Mia Michel El BuenonDiego MartinezBelum ada peringkat
- Reseña 17 Ingeleses Envenenados..Dokumen7 halamanReseña 17 Ingeleses Envenenados..Lucho VillamizarBelum ada peringkat