Anda mungkin juga menyukai
- Barriers To Pediatric Sickle Cell Disease Guideline Recommendations.Dokumen7 halamanBarriers To Pediatric Sickle Cell Disease Guideline Recommendations.arturomarticarvajalBelum ada peringkat
- Hooi Min Lim, Yook Chin Chia, Siew Mooi Ching, Karuthan ChinnaDokumen8 halamanHooi Min Lim, Yook Chin Chia, Siew Mooi Ching, Karuthan ChinnaarturomarticarvajalBelum ada peringkat
- The 9-Item Physician Documentation Quality Instrument (PDQI-9) Score Is Not Useful in Evaluating EMR (Scribe) Note Quality in Emergency Medicine.Dokumen13 halamanThe 9-Item Physician Documentation Quality Instrument (PDQI-9) Score Is Not Useful in Evaluating EMR (Scribe) Note Quality in Emergency Medicine.arturomarticarvajalBelum ada peringkat
- Isolated Propriospinal Myoclonus As A Presentation of Cervical Myelopathy.Dokumen2 halamanIsolated Propriospinal Myoclonus As A Presentation of Cervical Myelopathy.arturomarticarvajalBelum ada peringkat
- Targeted HIV Testing For Male Partners of HIV-positive Pregnant Women in A High Prevalence Setting in NigeriaDokumen12 halamanTargeted HIV Testing For Male Partners of HIV-positive Pregnant Women in A High Prevalence Setting in NigeriaarturomarticarvajalBelum ada peringkat
- Idcases: Stephanie Stephanie, Sarah A. SchmalzleDokumen3 halamanIdcases: Stephanie Stephanie, Sarah A. SchmalzlearturomarticarvajalBelum ada peringkat
- Sickle Cell Disease: Advances in TreatmentDokumen13 halamanSickle Cell Disease: Advances in TreatmentarturomarticarvajalBelum ada peringkat
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (400)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (588)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (266)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (74)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (121)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- Study 107 - The Doctrine of Salvation - Part 8Dokumen2 halamanStudy 107 - The Doctrine of Salvation - Part 8Jason MyersBelum ada peringkat
- Electro Fashion Sewable LED Kits WebDokumen10 halamanElectro Fashion Sewable LED Kits WebAndrei VasileBelum ada peringkat
- AIA1800 Operator ManualDokumen184 halamanAIA1800 Operator ManualZain Sa'adehBelum ada peringkat
- Presentation 11Dokumen14 halamanPresentation 11stellabrown535Belum ada peringkat
- Functional DesignDokumen17 halamanFunctional DesignRajivSharmaBelum ada peringkat
- MASONRYDokumen8 halamanMASONRYJowelyn MaderalBelum ada peringkat
- Opc PPT FinalDokumen22 halamanOpc PPT FinalnischalaBelum ada peringkat
- 3 ALCE Insulators 12R03.1Dokumen12 halaman3 ALCE Insulators 12R03.1Amílcar Duarte100% (1)
- Crown WF-3000 1.2Dokumen5 halamanCrown WF-3000 1.2Qirat KhanBelum ada peringkat
- Mixed Up MonstersDokumen33 halamanMixed Up MonstersjaneBelum ada peringkat
- Chemistry Form 4 Daily Lesson Plan - CompressDokumen3 halamanChemistry Form 4 Daily Lesson Plan - Compressadila ramlonBelum ada peringkat
- Fss Presentation Slide GoDokumen13 halamanFss Presentation Slide GoReinoso GreiskaBelum ada peringkat
- 5c3f1a8b262ec7a Ek PDFDokumen5 halaman5c3f1a8b262ec7a Ek PDFIsmet HizyoluBelum ada peringkat
- Carob-Tree As CO2 Sink in The Carbon MarketDokumen5 halamanCarob-Tree As CO2 Sink in The Carbon MarketFayssal KartobiBelum ada peringkat
- Kahneman & Tversky Origin of Behavioural EconomicsDokumen25 halamanKahneman & Tversky Origin of Behavioural EconomicsIan Hughes100% (1)
- Hamstring - WikipediaDokumen21 halamanHamstring - WikipediaOmar MarwanBelum ada peringkat
- Gods Omnipresence in The World On Possible MeaninDokumen20 halamanGods Omnipresence in The World On Possible MeaninJoan Amanci Casas MuñozBelum ada peringkat
- B122 - Tma03Dokumen7 halamanB122 - Tma03Martin SantambrogioBelum ada peringkat
- From Philo To Plotinus AftermanDokumen21 halamanFrom Philo To Plotinus AftermanRaphael888Belum ada peringkat
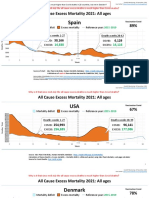
- Countries EXCESS DEATHS All Ages - 15nov2021Dokumen21 halamanCountries EXCESS DEATHS All Ages - 15nov2021robaksBelum ada peringkat
- Sam Media Recruitment QuestionnaireDokumen17 halamanSam Media Recruitment Questionnairechek taiBelum ada peringkat
- Read While Being Blind.. Braille's Alphabet: Be Aware and Active !Dokumen3 halamanRead While Being Blind.. Braille's Alphabet: Be Aware and Active !bitermanBelum ada peringkat
- The JHipster Mini Book 2Dokumen129 halamanThe JHipster Mini Book 2tyulist100% (1)
- Post Appraisal InterviewDokumen3 halamanPost Appraisal InterviewNidhi D100% (1)
- WEB DESIGN WITH AUSTINE-converted-1Dokumen9 halamanWEB DESIGN WITH AUSTINE-converted-1JayjayBelum ada peringkat
- Laboratory Manual (CIV 210) Engineering Surveying (2018-19) (For Private Circulation Only)Dokumen76 halamanLaboratory Manual (CIV 210) Engineering Surveying (2018-19) (For Private Circulation Only)gyanendraBelum ada peringkat
- Evaluation TemplateDokumen3 halamanEvaluation Templateapi-308795752Belum ada peringkat
- WL-80 FTCDokumen5 halamanWL-80 FTCMr.Thawatchai hansuwanBelum ada peringkat
- 13 Adsorption of Congo Red A Basic Dye by ZnFe-CO3Dokumen10 halaman13 Adsorption of Congo Red A Basic Dye by ZnFe-CO3Jorellie PetalverBelum ada peringkat
- A Case Study of Coustomer Satisfaction in Demat Account At: A Summer Training ReportDokumen110 halamanA Case Study of Coustomer Satisfaction in Demat Account At: A Summer Training ReportDeepak SinghalBelum ada peringkat