Anda mungkin juga menyukai
- Standard Operating Procedure Trial Master File and 421 Main Evidence in TheDokumen33 halamanStandard Operating Procedure Trial Master File and 421 Main Evidence in Theahmed.bouchenakBelum ada peringkat
- The Research ProtocolDokumen8 halamanThe Research Protocollil assBelum ada peringkat
- Screening and Enrollment: Standard Operating Procedures For Clinical Research at Kent State UniversityDokumen6 halamanScreening and Enrollment: Standard Operating Procedures For Clinical Research at Kent State UniversityAlberio CygnusBelum ada peringkat
- CLN-20 00 Investigational Product HandlingDokumen4 halamanCLN-20 00 Investigational Product Handlingpopatlilo2Belum ada peringkat
- Site Close-Out Visit: Standard Operating Procedures For Clinical Research at Kent State UniversityDokumen4 halamanSite Close-Out Visit: Standard Operating Procedures For Clinical Research at Kent State Universityna sarvaBelum ada peringkat
- SOP 303 SiteInitiationVisitDokumen3 halamanSOP 303 SiteInitiationVisitna sarvaBelum ada peringkat
- ScientificDokumen4 halamanScientificNarendar ReddyBelum ada peringkat
- Standard Operating Procedure for Source DocumentationDokumen29 halamanStandard Operating Procedure for Source DocumentationSreeraj Guruvayoor SBelum ada peringkat
- Nonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsDari EverandNonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsWilliam J. BrockBelum ada peringkat
- Medical Device Clinical Evaluation: Malesh MDokumen5 halamanMedical Device Clinical Evaluation: Malesh Myagnaashi9092100% (1)
- Infections Associated With Reprocessed Urological Endoscopes - Letter To Health Care Providers - FDADokumen3 halamanInfections Associated With Reprocessed Urological Endoscopes - Letter To Health Care Providers - FDAt merchant100% (1)
- Clinical Trial Agreements SOPDokumen6 halamanClinical Trial Agreements SOPInfo OptimusBTBelum ada peringkat
- Supplement 1 Medical Device Product QuestionnaireDokumen14 halamanSupplement 1 Medical Device Product QuestionnaireJi YuBelum ada peringkat
- Sop Title: SOP Version No: 01 Date:: Reviewing and Obtaining Informed ConsentDokumen12 halamanSop Title: SOP Version No: 01 Date:: Reviewing and Obtaining Informed ConsentMadhan MohanBelum ada peringkat
- Costing Methods For Clinical TrialsDokumen7 halamanCosting Methods For Clinical TrialsArun NairBelum ada peringkat
- SOP-QA-6 V4 Study Start-UpDokumen4 halamanSOP-QA-6 V4 Study Start-UpOlja PopovicBelum ada peringkat
- Budgeting For Clinical TrialsDokumen30 halamanBudgeting For Clinical TrialsArun NairBelum ada peringkat
- SOP 302 SiteQualificationVisitDokumen3 halamanSOP 302 SiteQualificationVisitna sarvaBelum ada peringkat
- 2a. Class A Compression DeviceDokumen31 halaman2a. Class A Compression DeviceCedric Bonneau100% (1)
- Clinical Study Report, Bioequivalence, General Concepts and OverviewDokumen48 halamanClinical Study Report, Bioequivalence, General Concepts and OverviewAhmad Abdullah Najjar100% (7)
- Archiving Trial DataDokumen10 halamanArchiving Trial DataArjun Singh ChoudharyBelum ada peringkat
- DePuy ASR Surgeon Recall PackDokumen10 halamanDePuy ASR Surgeon Recall PackKirkBernardBelum ada peringkat
- Sop Informed ConsentDokumen4 halamanSop Informed ConsentWelzha Meturan-kadmaerubunBelum ada peringkat
- Health Research Ethics Committees Human Research (HREC) : Standard Operating Procedures and Guidelines May 2010Dokumen38 halamanHealth Research Ethics Committees Human Research (HREC) : Standard Operating Procedures and Guidelines May 2010prabhaBelum ada peringkat
- Monitoring Plan and Standard Operating Procedure: Protocol TitleDokumen5 halamanMonitoring Plan and Standard Operating Procedure: Protocol TitleDipeshBelum ada peringkat
- CAP 1116 USP Control de AmbientesDokumen14 halamanCAP 1116 USP Control de AmbientesCamilo Florez100% (1)
- Introduction Clinical TrialsDokumen22 halamanIntroduction Clinical TrialsSharadBelum ada peringkat
- Content: Standard Operating Procedure (SOP) Master SOP For Writing Quality Management DocumentsDokumen6 halamanContent: Standard Operating Procedure (SOP) Master SOP For Writing Quality Management DocumentsAmer RahmahBelum ada peringkat
- SOP Formats ManagementDokumen5 halamanSOP Formats ManagementJayant Kumar JhaBelum ada peringkat
- Facility Validation: A New Approach: Process InvolvementDokumen4 halamanFacility Validation: A New Approach: Process InvolvementsukmaBelum ada peringkat
- Adverse Drug Reaction FormDokumen2 halamanAdverse Drug Reaction FormDhananjay Saini100% (1)
- WWORTH SOP10ProjectManagementV2.2 140507Dokumen21 halamanWWORTH SOP10ProjectManagementV2.2 140507Fery Alapola100% (1)
- BT750 - Clinical Evaluation ReportDokumen45 halamanBT750 - Clinical Evaluation Reportfjvillamunoz100% (1)
- SopDokumen3 halamanSopmrchilliciousBelum ada peringkat
- Standard Operating Procedure: TitleDokumen9 halamanStandard Operating Procedure: TitleayodejidetaBelum ada peringkat
- GHTF Sg5 n4 Post Market Clinical Studies 100218Dokumen10 halamanGHTF Sg5 n4 Post Market Clinical Studies 100218India RoseBelum ada peringkat
- Applicable Standards and Common SpecificationsDokumen4 halamanApplicable Standards and Common Specificationsaymohamad2020Belum ada peringkat
- Clinical Trial ChecklistDokumen3 halamanClinical Trial ChecklistShruti CholaBelum ada peringkat
- NIDCR Clinical TrialDokumen67 halamanNIDCR Clinical TrialEnrique TrvjilloBelum ada peringkat
- MedDRA N SOPDokumen32 halamanMedDRA N SOPsuri33350% (2)
- MDR New Update GeneralDokumen6 halamanMDR New Update Generalpramod baghelBelum ada peringkat
- EU Tech File White Paper EmergoDokumen8 halamanEU Tech File White Paper Emergogobu269104Belum ada peringkat
- Joint Research Arrangements SOPDokumen9 halamanJoint Research Arrangements SOPmo2bioBelum ada peringkat
- MDCG 2020 - 5 Clinical Evaluation - Equivalence - April 2020Dokumen20 halamanMDCG 2020 - 5 Clinical Evaluation - Equivalence - April 2020Kangjin JeonBelum ada peringkat
- Medical Device Reporting Regulation and Electronic SubmissionDokumen26 halamanMedical Device Reporting Regulation and Electronic Submissionvinaysetty100% (1)
- GCP Compliance QuestionsDokumen2 halamanGCP Compliance QuestionsJaime HinojosaBelum ada peringkat
- 2 - 7-1 - 04-2003 Clinical EvaluationDokumen20 halaman2 - 7-1 - 04-2003 Clinical EvaluationGABYBelum ada peringkat
- Risk Management Report: Mapping of Standard Requirements To Document SectionsDokumen3 halamanRisk Management Report: Mapping of Standard Requirements To Document SectionsSIS AuditorBelum ada peringkat
- Protocol Template 05feb2016 508Dokumen61 halamanProtocol Template 05feb2016 508Dawit Birhanu100% (1)
- BACR Presentation GCP Jan-2018-002Dokumen72 halamanBACR Presentation GCP Jan-2018-002MilenBelum ada peringkat
- TEM 300 Product Transfer Protocol Template SampleDokumen7 halamanTEM 300 Product Transfer Protocol Template SampleamebadhaBelum ada peringkat
- Scalpel Safety Sinnott2010Dokumen9 halamanScalpel Safety Sinnott2010Guille Puertas100% (1)
- NDA ProcessDokumen3 halamanNDA Processdustymarie100% (2)
- SOP Template For Preparation of A Clinical Trial Authorisation V4 1Dokumen5 halamanSOP Template For Preparation of A Clinical Trial Authorisation V4 1DrSyeda Rima100% (1)
- SITE GEN-001 01 - SOP On Site ActivitiesDokumen17 halamanSITE GEN-001 01 - SOP On Site ActivitiesCR Professionals IndiaBelum ada peringkat
- Meddev 2.7 4Dokumen10 halamanMeddev 2.7 4Jug_HustlerBelum ada peringkat
- Tem-300 Product Transfer ProtocolDokumen15 halamanTem-300 Product Transfer Protocolthien Doan100% (1)
- Medical Devices - SOP 3 - Acceptance of Medical DevicesDokumen6 halamanMedical Devices - SOP 3 - Acceptance of Medical DevicesCaalaa Dabalaa LamuuBelum ada peringkat
- Suspected Adverse Reactions Form 0Dokumen1 halamanSuspected Adverse Reactions Form 0markBelum ada peringkat
- Data Integrity and Compliance: A Primer for Medical Product ManufacturersDari EverandData Integrity and Compliance: A Primer for Medical Product ManufacturersBelum ada peringkat
- Real-Time PCR Automations of Quant Studio 5 and MA6000 PlusDokumen15 halamanReal-Time PCR Automations of Quant Studio 5 and MA6000 PlusDayledaniel SorvetoBelum ada peringkat
- Anatomy and Physiology Lecture DiscussionDokumen27 halamanAnatomy and Physiology Lecture DiscussionDayledaniel SorvetoBelum ada peringkat
- Real-Time PCR Automations of Quant Studio 5 and MA6000 PlusDokumen15 halamanReal-Time PCR Automations of Quant Studio 5 and MA6000 PlusDayledaniel SorvetoBelum ada peringkat
- PMLS LAB EXERCISE IN Venipuncture ProceduresDokumen3 halamanPMLS LAB EXERCISE IN Venipuncture ProceduresDayledaniel SorvetoBelum ada peringkat
- PMLS LAB EXERCISE IN Venipuncture ProceduresDokumen3 halamanPMLS LAB EXERCISE IN Venipuncture ProceduresDayledaniel SorvetoBelum ada peringkat
- Real-Time PCR Automations of Quant Studio 5 and MA6000 PlusDokumen15 halamanReal-Time PCR Automations of Quant Studio 5 and MA6000 PlusDayledaniel SorvetoBelum ada peringkat
- Anaerobe of Clinical ImportanceDokumen43 halamanAnaerobe of Clinical ImportanceDayledaniel SorvetoBelum ada peringkat
- ANAPHY Disease PathologyDokumen19 halamanANAPHY Disease PathologyDayledaniel SorvetoBelum ada peringkat
- Non Hodgkin's Lymphoma: Rakesh BiswasDokumen16 halamanNon Hodgkin's Lymphoma: Rakesh BiswasDayledaniel SorvetoBelum ada peringkat
- Water, PH, and Ionic Equilibria: To Accompany Biochemistry, 2/e by Reginald Garrett and Charles GrishamDokumen32 halamanWater, PH, and Ionic Equilibria: To Accompany Biochemistry, 2/e by Reginald Garrett and Charles GrishamRoshin TejeroBelum ada peringkat
- Case Study Analysis (1,2,3,4) ENDOCRINOLOGYDokumen3 halamanCase Study Analysis (1,2,3,4) ENDOCRINOLOGYDayledaniel Sorveto100% (1)
- InversionSucrose IngleDokumen10 halamanInversionSucrose IngleAida Alonso MartinezBelum ada peringkat
- Proteins: Prepared By: Dayle Daniel G. Sorveto, RMT, MSMTDokumen64 halamanProteins: Prepared By: Dayle Daniel G. Sorveto, RMT, MSMTDayledaniel SorvetoBelum ada peringkat
- Chapter 33: Protein SynthesisDokumen64 halamanChapter 33: Protein SynthesisDayledaniel SorvetoBelum ada peringkat
- Activity - Body TissuesDokumen8 halamanActivity - Body TissuesDayledaniel SorvetoBelum ada peringkat
- Blood Vessel and Lymphatic Lab OutputDokumen12 halamanBlood Vessel and Lymphatic Lab OutputDayledaniel SorvetoBelum ada peringkat
- ProteinsDokumen64 halamanProteinsDayledaniel SorvetoBelum ada peringkat
- Week 1 Pmls2Dokumen45 halamanWeek 1 Pmls2Dayledaniel SorvetoBelum ada peringkat
- Week 3 Cellular Levels of Organization OutputDokumen8 halamanWeek 3 Cellular Levels of Organization OutputDayledaniel SorvetoBelum ada peringkat
- Lab Output Anaphy 6-Integumentary SystemDokumen6 halamanLab Output Anaphy 6-Integumentary SystemDayledaniel SorvetoBelum ada peringkat
- Heart Lab OutputDokumen10 halamanHeart Lab OutputDayledaniel Sorveto100% (1)
- Comparison of Connective Tissues Types and FunctionsDokumen1 halamanComparison of Connective Tissues Types and FunctionsDayledaniel SorvetoBelum ada peringkat
- Anatomy and Physiology Chapter11 The Muscular SystemDokumen18 halamanAnatomy and Physiology Chapter11 The Muscular SystemDayledaniel SorvetoBelum ada peringkat
- Endocrine SystemDokumen11 halamanEndocrine SystemDayledaniel SorvetoBelum ada peringkat
- Chapter 13 highlights key aspects of bloodDokumen8 halamanChapter 13 highlights key aspects of bloodDayledaniel Sorveto0% (1)
- Ch. 1 IntroductionDokumen39 halamanCh. 1 IntroductionPiao Liang JingBelum ada peringkat
- Lecciones Basicas en Control de Calidad.2008 Bio-Rad InglesDokumen62 halamanLecciones Basicas en Control de Calidad.2008 Bio-Rad InglesPili Gabriel67% (3)
- Analysis of Urine and Other Body Fluids (WLP Draft)Dokumen1 halamanAnalysis of Urine and Other Body Fluids (WLP Draft)Dayledaniel SorvetoBelum ada peringkat
- Skin Model ProjectDokumen2 halamanSkin Model ProjectDayledaniel SorvetoBelum ada peringkat
- Case Studies in Bacteriology Legaspi KimDokumen9 halamanCase Studies in Bacteriology Legaspi KimDayledaniel SorvetoBelum ada peringkat
- GEH-6680LCI FaultsDokumen76 halamanGEH-6680LCI FaultsMuhammad IdreesarainBelum ada peringkat
- Euro 4 Standard PDFDokumen2 halamanEuro 4 Standard PDFCamellaBelum ada peringkat
- Dr. Berg's Favorite HEALTHY JUNK FOODS & Other AlternativesDokumen23 halamanDr. Berg's Favorite HEALTHY JUNK FOODS & Other Alternativesprashant_padte100% (8)
- Indg 449Dokumen12 halamanIndg 449Nissam SidheeqBelum ada peringkat
- Catalogo Unidad Enfriadora Trane R-407C PDFDokumen8 halamanCatalogo Unidad Enfriadora Trane R-407C PDFJUAN FRANCISCO AYALABelum ada peringkat
- BOD FormatDokumen4 halamanBOD FormatSkill IndiaBelum ada peringkat
- Airborne Life Support Systems In-House Infant Transport Sys 185A Infant Incubator - Manufacturer SpecificationsDokumen2 halamanAirborne Life Support Systems In-House Infant Transport Sys 185A Infant Incubator - Manufacturer SpecificationsAsistir Biomedica SASBelum ada peringkat
- Lecture 1 Biochem 232 CellsDokumen13 halamanLecture 1 Biochem 232 CellsaelmowafyBelum ada peringkat
- Egg Pasteurization Manual 1969Dokumen54 halamanEgg Pasteurization Manual 1969Tomas MuzzioBelum ada peringkat
- ABB Leaflet Comem BR-En 2018-06-07Dokumen2 halamanABB Leaflet Comem BR-En 2018-06-07Dave ChaudhuryBelum ada peringkat
- ABS Part4 - Vessel Systems & Machinery - 2001Dokumen710 halamanABS Part4 - Vessel Systems & Machinery - 2001AndréMenezesBelum ada peringkat
- 6.water Treatment and Make-Up Water SystemDokumen18 halaman6.water Treatment and Make-Up Water Systempepenapao1217100% (1)
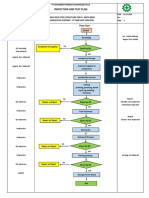
- Inspection and Test Plan: Flow Chart Start IncomingDokumen1 halamanInspection and Test Plan: Flow Chart Start IncomingSinden AyuBelum ada peringkat
- Final TLE9 Nail Care9 Q1 Module 3Dokumen20 halamanFinal TLE9 Nail Care9 Q1 Module 3Ma. Andrea LagmanBelum ada peringkat
- Sampling & Data AssayDokumen22 halamanSampling & Data AssayFerdinand SiahaanBelum ada peringkat
- SmithfieldDokumen11 halamanSmithfieldandreea143Belum ada peringkat
- ENGLISH TEST 9 SMPDokumen4 halamanENGLISH TEST 9 SMPMariatul afniBelum ada peringkat
- Variable Displacement Engines: The Magic of Cylinder DeactivationDokumen3 halamanVariable Displacement Engines: The Magic of Cylinder DeactivationdinuBelum ada peringkat
- Experiment Report Basic Physics "Total Internal Reflection"Dokumen10 halamanExperiment Report Basic Physics "Total Internal Reflection"dita wulanBelum ada peringkat
- Asme NM.2-2018Dokumen126 halamanAsme NM.2-2018aneeshjokay75% (4)
- Schneider - Cptg010 en (Print)Dokumen16 halamanSchneider - Cptg010 en (Print)el_koptan00857693Belum ada peringkat
- Fischer FBN II BoltDokumen5 halamanFischer FBN II BoltJaga NathBelum ada peringkat
- Generate Profits from Bottled Water Using Atmospheric Water GeneratorsDokumen20 halamanGenerate Profits from Bottled Water Using Atmospheric Water GeneratorsJose AndradeBelum ada peringkat
- Acute Coronary Syndrome Guidelines 2020Dokumen79 halamanAcute Coronary Syndrome Guidelines 2020Γιώργος ΕλευθεριάδηςBelum ada peringkat
- Edinburgh Postnatal Depression Scale. Detection of Postnatal Depression. Development of The 10-ItemDokumen6 halamanEdinburgh Postnatal Depression Scale. Detection of Postnatal Depression. Development of The 10-ItemKyze LQBelum ada peringkat
- Informática Ejercicios IDokumen10 halamanInformática Ejercicios IAlejandroMendezBelum ada peringkat
- Specialized Connective TissueDokumen15 halamanSpecialized Connective TissueSebBelum ada peringkat
- Lesson 5 - The Problem of EvilDokumen10 halamanLesson 5 - The Problem of Evilsemmerson4896Belum ada peringkat
- Health Programs Activities Timeframe Expected Output Child CareDokumen3 halamanHealth Programs Activities Timeframe Expected Output Child CareC SamBelum ada peringkat
- Darnell's Father Goes StrictDokumen2 halamanDarnell's Father Goes StrictDavid Theodore Richardson IIIBelum ada peringkat