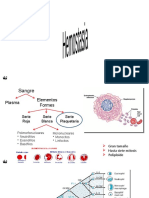
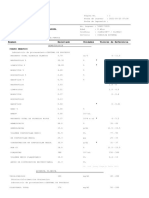
Anda mungkin juga menyukai
- Trombastenia de GlazmannDokumen13 halamanTrombastenia de Glazmanncarlina rivasBelum ada peringkat
- La Tromboastenia de GlanzmannDokumen2 halamanLa Tromboastenia de GlanzmannFiorella Aguilar ValenciaBelum ada peringkat
- Práctica 4 Pruebas de CoágulaciónDokumen11 halamanPráctica 4 Pruebas de CoágulaciónSandy MoralesBelum ada peringkat
- Aclaramiento de CreatininaDokumen2 halamanAclaramiento de CreatininaCamilaApoloTorres100% (1)
- Resumen Sistema AboDokumen3 halamanResumen Sistema AboAnonymous 5RhHmNmgJBelum ada peringkat
- Informe AnticoagulantesDokumen27 halamanInforme AnticoagulantesJavieraPssBelum ada peringkat
- Proyecto VSGDokumen29 halamanProyecto VSGOctavio VillegasBelum ada peringkat
- Inmuno-Hematología y Medicina TransfusionalDokumen40 halamanInmuno-Hematología y Medicina TransfusionalGabriela100% (1)
- Práctica de LeucogramaDokumen7 halamanPráctica de LeucogramaAsael VzBelum ada peringkat
- Fraccionamiento de La SangreDokumen3 halamanFraccionamiento de La Sangrezea100% (1)
- Trombocitopenia Inducida Por FármacosDokumen14 halamanTrombocitopenia Inducida Por FármacosLuis Andrade100% (1)
- La Diferencia Entre Plasma y SueroDokumen1 halamanLa Diferencia Entre Plasma y SueroSebastian ToroBelum ada peringkat
- Médula Médula Suprarrenal Suprarrenal: Salgado Valenzuela Cinthya FernandaDokumen34 halamanMédula Médula Suprarrenal Suprarrenal: Salgado Valenzuela Cinthya FernandaMarco Alejandro Guerra MorenoBelum ada peringkat
- Informe de Liquido AsciticoDokumen6 halamanInforme de Liquido AsciticoRosa Merino FernandezBelum ada peringkat
- Perfil hepático: guía para interpretar exámenesDokumen7 halamanPerfil hepático: guía para interpretar exámenesStephanyChavezFeriaBelum ada peringkat
- 2012.valores de VSG Por El Metodo de Westergren Con Dispette Diluido y Sin DiluirDokumen22 halaman2012.valores de VSG Por El Metodo de Westergren Con Dispette Diluido y Sin DiluirMarianela AlzuruBelum ada peringkat
- Evaluacion Analitica Del Metabolismo de Carbohidratos: Tecnólogo Médico Esp. Laboratorio Clínico y Anatomía PatológicaDokumen64 halamanEvaluacion Analitica Del Metabolismo de Carbohidratos: Tecnólogo Médico Esp. Laboratorio Clínico y Anatomía Patológicavalentinatay100% (1)
- Reacciones FebrilesDokumen24 halamanReacciones FebrilesMaria HuertaBelum ada peringkat
- Hemocultivo Practicas Microbiologia BiologiaDokumen3 halamanHemocultivo Practicas Microbiologia BiologiaOmarElmxSucasacaBelum ada peringkat
- 3 - Fisiologia de La Sangre - Hemostasia - 2014-IDokumen46 halaman3 - Fisiologia de La Sangre - Hemostasia - 2014-IDharyll Chancasanampa NarvaezBelum ada peringkat
- Química Del Proceso de FijaciónDokumen52 halamanQuímica Del Proceso de FijaciónAdolfo Antonio Ríos AlcortaBelum ada peringkat
- 15 16 BANCO DE SANGRE y REACCIONES TRANSFUSIONALESDokumen161 halaman15 16 BANCO DE SANGRE y REACCIONES TRANSFUSIONALESAndres TuestaBelum ada peringkat
- Perfil CardíacoDokumen7 halamanPerfil CardíacoDiana JuradoBelum ada peringkat
- G.rojos Normales y AnormalesDokumen51 halamanG.rojos Normales y AnormalesDay Blaakee0% (1)
- Perfil de CoagulaciónDokumen2 halamanPerfil de CoagulaciónTatiana RamirezBelum ada peringkat
- Sindrome Anemico y PurpuricoDokumen28 halamanSindrome Anemico y PurpuricoGuillermo Pacheco RivasBelum ada peringkat
- GalactosuriaDokumen21 halamanGalactosuriaErcelyn Menjivar50% (2)
- Anticoagulante Lúpico 2Dokumen17 halamanAnticoagulante Lúpico 2Alison Nicol100% (1)
- Aglt y PostzonaDokumen34 halamanAglt y Postzonayleys83% (6)
- Anticuerpo AntiplaquetariosDokumen18 halamanAnticuerpo Antiplaquetarioslourdes antonia checca escobedo100% (1)
- Prueba de La OptoquinaDokumen1 halamanPrueba de La OptoquinaOscar Chuquiyauri PomacinoBelum ada peringkat
- Ciclo de Plasmodium: esporozoito a gametocitoDokumen2 halamanCiclo de Plasmodium: esporozoito a gametocitoeduardoBelum ada peringkat
- Fibrinogeno SPDokumen3 halamanFibrinogeno SPandreaBelum ada peringkat
- Tema 1 HemostasiaDokumen127 halamanTema 1 HemostasiaBrigitteMoranBelum ada peringkat
- Hibridación de Ácidos NucleicosDokumen34 halamanHibridación de Ácidos NucleicosDiana Camacho PlascenciaBelum ada peringkat
- 11.1 CoproanalisisDokumen35 halaman11.1 CoproanalisisMarian GarciaBelum ada peringkat
- Tecnicas InmunologicasDokumen34 halamanTecnicas InmunologicasQcarlos GroTpBelum ada peringkat
- Sistema de Coagulación y CininasDokumen2 halamanSistema de Coagulación y CininasFresya MejiaBelum ada peringkat
- Activación de Linfocitos TDokumen3 halamanActivación de Linfocitos TOctavio ZmBelum ada peringkat
- Anatomia Patologica. Casos ClinicosDokumen7 halamanAnatomia Patologica. Casos ClinicosGABRIELAJGRBelum ada peringkat
- Citometría HemáticaDokumen14 halamanCitometría HemáticaJoel Zuñiga RoblesBelum ada peringkat
- 25 Perfil LipídicoDokumen6 halaman25 Perfil LipídicoJose AragonBelum ada peringkat
- Conteo de EritrocitosDokumen3 halamanConteo de EritrocitosReinaldo Joel CerdanBelum ada peringkat
- Albumina Aa SPDokumen9 halamanAlbumina Aa SPasd pvpBelum ada peringkat
- Practica de MariaDokumen18 halamanPractica de MariaChavez PoolBelum ada peringkat
- Frotis de Sangre PeriféricaDokumen21 halamanFrotis de Sangre PeriféricaIssaBarreraBelum ada peringkat
- Clase 4 Funcion RenalDokumen49 halamanClase 4 Funcion RenalJaz Ntvg100% (1)
- Laboratorio DilucionesDokumen4 halamanLaboratorio DilucionesValeria Hernandez100% (1)
- Penicilinas. Utilidad y LimitacionesDokumen36 halamanPenicilinas. Utilidad y LimitacionesErick Efrain Carlo OtaloraBelum ada peringkat
- Anemias hemolíticas inmunes: tipos y mecanismosDokumen20 halamanAnemias hemolíticas inmunes: tipos y mecanismosChristian Cuello MendozaBelum ada peringkat
- VDRLDokumen2 halamanVDRLOscar NickBelum ada peringkat
- Tiempo de Lisis de EuglobulinasDokumen2 halamanTiempo de Lisis de EuglobulinasFloydRushBelum ada peringkat
- Ejercicio de Casos Clínicos de Micosis OportunistasDokumen12 halamanEjercicio de Casos Clínicos de Micosis OportunistasdarkblkBelum ada peringkat
- IndometacinaDokumen8 halamanIndometacinaLeonardo AlBruzBelum ada peringkat
- Técnicas para Determinar ElectrolitosDokumen2 halamanTécnicas para Determinar ElectrolitosAlfonso Hernandez0% (1)
- SX Bernard-Soulier y Tromboastenia GlicoproteínasDokumen8 halamanSX Bernard-Soulier y Tromboastenia GlicoproteínasBri Gonzalez TrejoBelum ada peringkat
- Trombastenia de GlanzmannDokumen5 halamanTrombastenia de GlanzmannDANNA JENDAY MARTINEZ POSADASBelum ada peringkat
- Lectura Trastornos Cualitat de Plaquetas PDFDokumen12 halamanLectura Trastornos Cualitat de Plaquetas PDFAngel CruzBelum ada peringkat
- Informe Seminario Taller #04 - Subgrupo 5Dokumen6 halamanInforme Seminario Taller #04 - Subgrupo 5Brayan Pinedo RodriguezBelum ada peringkat
- Trabajo Síndrome de Bernard SoulierDokumen5 halamanTrabajo Síndrome de Bernard SoulierLorenz Celeste CitalanBelum ada peringkat
- Resultado 1044433165 010463733751WvlJ 0 0FIDokumen1 halamanResultado 1044433165 010463733751WvlJ 0 0FIjose jimenezBelum ada peringkat
- Línea Del Tiempo EmbriológicaDokumen1 halamanLínea Del Tiempo EmbriológicaAbigail Alonso FranciscoBelum ada peringkat
- Aplicación Del PRP y Del PDGF en Periodoncia. Revisión de La LiteraturaDokumen8 halamanAplicación Del PRP y Del PDGF en Periodoncia. Revisión de La LiteraturaPedro Tomas Hernandez GuarimanBelum ada peringkat
- Exposiciones MOF110 1ra ParteDokumen2 halamanExposiciones MOF110 1ra ParteFP MarcoBelum ada peringkat
- Anemia Células Falciformes: Causas, Síntomas y TratamientoDokumen43 halamanAnemia Células Falciformes: Causas, Síntomas y TratamientoMilagros Guzmán GarcíaBelum ada peringkat
- Anemia Hemolitica AutoinmuneDokumen11 halamanAnemia Hemolitica AutoinmunemariBelum ada peringkat
- Sistemaseo 101030163942 Phpapp01Dokumen14 halamanSistemaseo 101030163942 Phpapp01Nilreyam FerBelum ada peringkat
- Hematologia AviarDokumen11 halamanHematologia AviarMagaly JuBa Vampilita AkiusBelum ada peringkat
- Clase 4 - Sistema NeuroMuscularDokumen32 halamanClase 4 - Sistema NeuroMuscularAyelen SchwabBelum ada peringkat
- Cuestionario HistologíaDokumen11 halamanCuestionario HistologíaEverardo MartinezBelum ada peringkat
- FlebotomíaDokumen3 halamanFlebotomíaJessica Juditt De Los Santos PaimaBelum ada peringkat
- CianocrilatoDokumen6 halamanCianocrilatoJudith Huarancca MariñoBelum ada peringkat
- Enfermedad de Von WillembrandDokumen2 halamanEnfermedad de Von WillembrandIsabela Rosero CastroBelum ada peringkat
- INFORME 9 BiologíaDokumen8 halamanINFORME 9 BiologíaZully VilchezBelum ada peringkat
- Tejido ÓseoDokumen7 halamanTejido ÓseodanielaBelum ada peringkat
- Actividad2 Vergara AdolfoDokumen7 halamanActividad2 Vergara AdolfoAdolfo Vergara MejíaBelum ada peringkat
- Valores normales-BHDokumen1 halamanValores normales-BHHelios EspinosaBelum ada peringkat
- Examen Laboratorio - 2021052501203Dokumen4 halamanExamen Laboratorio - 2021052501203Yury Andrea Busto ValeroBelum ada peringkat
- Valores Normales - Examenes de LaboratorioDokumen8 halamanValores Normales - Examenes de Laboratoriogabizuv100% (1)
- 01 Interpretación Del Hemograma en PediatríaDokumen24 halaman01 Interpretación Del Hemograma en PediatríaconimedinasBelum ada peringkat
- IntroducciónDokumen9 halamanIntroducciónRafael Hote Amador BlancoBelum ada peringkat
- Cuadro ComparativoDokumen6 halamanCuadro ComparativoJhon Alexander RODRIGUEZ SOTELOBelum ada peringkat
- Interpretacion Del HemogramaDokumen46 halamanInterpretacion Del HemogramaSara Soria EstrugoBelum ada peringkat
- RP001Dokumen3 halamanRP001natalia muñozBelum ada peringkat
- Celulas RojasDokumen3 halamanCelulas RojasANGIE JOHANNY HUAMANI PORTOCARREROBelum ada peringkat
- Anat. (01) Tejido Epitelial (I) 33 - 38Dokumen6 halamanAnat. (01) Tejido Epitelial (I) 33 - 38Angielizbeth RomerocarranzaBelum ada peringkat
- HematologíaDokumen2 halamanHematologíaOrnellaB0% (1)
- Hematologia: Cuadro HematicoDokumen1 halamanHematologia: Cuadro Hematiconatanael abreo chaparroBelum ada peringkat
- Concentrados de plaquetas: indicaciones y contraindicacionesDokumen6 halamanConcentrados de plaquetas: indicaciones y contraindicacionesIncomprendido Wilmer0% (1)
- Foro OsteologíaDokumen2 halamanForo OsteologíaEsther PizanoBelum ada peringkat