Anda mungkin juga menyukai
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (894)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (587)
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (119)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- 380 Final PaperDokumen46 halaman380 Final Paperapi-538048965Belum ada peringkat
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2219)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- Complaints Handling: BDA AdviceDokumen8 halamanComplaints Handling: BDA Advicedruzair007Belum ada peringkat
- Carlin Smith Child Abuse FinalDokumen12 halamanCarlin Smith Child Abuse FinalCarlin SmithBelum ada peringkat
- S. No Name and Address Form 25 Dated Validupto Form 28 Dated Validupto 1 List of Pharmaceuticals Manufacturing CompaniesDokumen52 halamanS. No Name and Address Form 25 Dated Validupto Form 28 Dated Validupto 1 List of Pharmaceuticals Manufacturing CompaniesSimon YiuBelum ada peringkat
- OPINION ESSAY AbortionDokumen2 halamanOPINION ESSAY AbortionLuisa Patiño100% (1)
- Food Safety 3Dokumen22 halamanFood Safety 3Syarahdita Fransiska RaniBelum ada peringkat
- Manu SkripDokumen20 halamanManu SkripzanimarBelum ada peringkat
- hdf492 Portfolio PresentationDokumen14 halamanhdf492 Portfolio Presentationapi-403412647Belum ada peringkat
- Nursing Assignment SampleDokumen12 halamanNursing Assignment Sampleswetha swethaBelum ada peringkat
- Things of Boundaries. Andrew AbbottDokumen27 halamanThings of Boundaries. Andrew AbbottDaniel SotoBelum ada peringkat
- NCERT Solutions For Class 7 Science Chapter 2Dokumen5 halamanNCERT Solutions For Class 7 Science Chapter 2SANJEEV KUMARBelum ada peringkat
- 3D Printing Technology in Drug Delivery: Recent Progress and ApplicationDokumen10 halaman3D Printing Technology in Drug Delivery: Recent Progress and ApplicationAngela DelarmenteBelum ada peringkat
- Class 7 PolityDokumen10 halamanClass 7 PolityNakka nikithaBelum ada peringkat
- Psychoeducational and Family Therapy in Relapse PreventionDokumen4 halamanPsychoeducational and Family Therapy in Relapse PreventionEdson HilárioBelum ada peringkat
- Email chat about sex actsDokumen10 halamanEmail chat about sex actsrishav kumar agarwalBelum ada peringkat
- 4020 Assessment 4 Instructions - Improvement Plan Tool Kit - ..Dokumen4 halaman4020 Assessment 4 Instructions - Improvement Plan Tool Kit - ..Sabahat BashirBelum ada peringkat
- Effects of Vitamin B-12 Supplementation On Neurologic and Cognitive Function in Older People: A Randomized Controlled TrialDokumen9 halamanEffects of Vitamin B-12 Supplementation On Neurologic and Cognitive Function in Older People: A Randomized Controlled TrialzuzuoonBelum ada peringkat
- STP 1560-2013Dokumen346 halamanSTP 1560-2013HieuHTBelum ada peringkat
- Jha For Painting of EquipmentDokumen1 halamanJha For Painting of EquipmentShahid RazaBelum ada peringkat
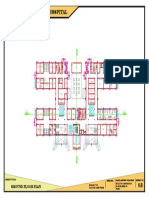
- 100-Bed General Hospital LayoutDokumen1 halaman100-Bed General Hospital LayoutAshish chauhanBelum ada peringkat
- Jacked at Home - Bodyweight Muscle-Building WorkoutsDokumen6 halamanJacked at Home - Bodyweight Muscle-Building Workoutsmohamed aliBelum ada peringkat
- Tushar FinalDokumen29 halamanTushar FinalRaj Prixit RathoreBelum ada peringkat
- Come Back To Your Senses Use Your Body: Psychologyt LsDokumen1 halamanCome Back To Your Senses Use Your Body: Psychologyt LsMarina Moran100% (1)
- K55 MSDSDokumen7 halamanK55 MSDSalocBelum ada peringkat
- Paul B. Bishop, DC, MD, PHD, Jeffrey A. Quon, DC, PHD, FCCSC, Charles G. Fisher, MD, MHSC, FRCSC, Marcel F.S. Dvorak, MD, FRCSCDokumen10 halamanPaul B. Bishop, DC, MD, PHD, Jeffrey A. Quon, DC, PHD, FCCSC, Charles G. Fisher, MD, MHSC, FRCSC, Marcel F.S. Dvorak, MD, FRCSCorlando moraBelum ada peringkat
- MANAGEMENT AND PREVENTIONDokumen6 halamanMANAGEMENT AND PREVENTIONIrina BalutaBelum ada peringkat
- 10 Doh Approved Herbal MedicineDokumen32 halaman10 Doh Approved Herbal MedicineIsmael JaaniBelum ada peringkat
- PORNOGRAPHICDokumen13 halamanPORNOGRAPHICcarlos ortizBelum ada peringkat
- A Medical Outreach Elective CourseDokumen11 halamanA Medical Outreach Elective CourseRobert SmithBelum ada peringkat
- Case Study - Genetic DisordersDokumen3 halamanCase Study - Genetic Disordersapi-340003532100% (1)