Anda mungkin juga menyukai
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (894)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (265)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (119)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- Fresh Gas DecouplingDokumen13 halamanFresh Gas DecouplingDo NodsBelum ada peringkat
- 9100c NXT: The Anesthesia Workstation That Gives You Peace of MindDokumen2 halaman9100c NXT: The Anesthesia Workstation That Gives You Peace of MindABHINANDAN SHARMA33% (3)
- GrassinoDokumen107 halamanGrassinomilelytaBelum ada peringkat
- Management of Pneumothorax With Oxygen Therapy A CDokumen4 halamanManagement of Pneumothorax With Oxygen Therapy A CAndreas Paian SiahaanBelum ada peringkat
- Be Great - Ebook of 365 Inspirational QuotesDokumen192 halamanBe Great - Ebook of 365 Inspirational Quotesnagesh2013Belum ada peringkat
- Photosynthesis and Cellular Respiration Test AP BioDokumen7 halamanPhotosynthesis and Cellular Respiration Test AP Bioafbsyebpu100% (1)
- Respiratory System: Study: Pulmonology Physician: Pulmonologist Function: Exchange of GasesDokumen5 halamanRespiratory System: Study: Pulmonology Physician: Pulmonologist Function: Exchange of GasesSakshi BishnoiBelum ada peringkat
- Obstructive Versus Restrictive Pulmonary DiseaseDokumen47 halamanObstructive Versus Restrictive Pulmonary DiseaseAntony WaithakaBelum ada peringkat
- Disturbances in Respiratory FunctionDokumen6 halamanDisturbances in Respiratory FunctionSeff CausapinBelum ada peringkat
- CopdandexerciseDokumen11 halamanCopdandexerciseapi-663023028Belum ada peringkat
- Breathing ProcessDokumen7 halamanBreathing ProcessJasmineBelum ada peringkat
- Concept of Disease 1.1 Anatomy and PhysiologyDokumen15 halamanConcept of Disease 1.1 Anatomy and PhysiologyAulia SandraBelum ada peringkat
- Chest Physiotherapy: NCM 106 LEC DEMO (Leininger)Dokumen27 halamanChest Physiotherapy: NCM 106 LEC DEMO (Leininger)coosa liquorsBelum ada peringkat
- COPD Case StudyDokumen6 halamanCOPD Case Studyvanessa100% (1)
- Respiratory System Organs and FunctionsDokumen8 halamanRespiratory System Organs and FunctionsJoselito JardielBelum ada peringkat
- Apnea of Prematurity: UnderstandingDokumen2 halamanApnea of Prematurity: UnderstandingEmil JaniakBelum ada peringkat
- Respiratory Assessment TheoryDokumen23 halamanRespiratory Assessment TheoryPopa SimonaBelum ada peringkat
- Test Bank For Pilbeams Mechanical Ventilation 6th Edition by CairoDokumen9 halamanTest Bank For Pilbeams Mechanical Ventilation 6th Edition by Cairochowryurduq0krhBelum ada peringkat
- Chapter 5: Major Metabolic Pathways: David Shonnard Department of Chemical Engineering Michigan Technological UniversityDokumen17 halamanChapter 5: Major Metabolic Pathways: David Shonnard Department of Chemical Engineering Michigan Technological UniversityLTE002Belum ada peringkat
- Clinical Teaching on Mechanical VentilatorDokumen24 halamanClinical Teaching on Mechanical VentilatorGiri SivaBelum ada peringkat
- Human Diseases Case Study 19CDokumen3 halamanHuman Diseases Case Study 19Cairickann50% (2)
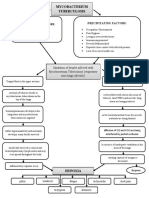
- Mycobacterium Tuberculosis: Precipitating Factors: Predisposing FactorsDokumen1 halamanMycobacterium Tuberculosis: Precipitating Factors: Predisposing FactorsYoko Mae Yano100% (1)
- Atelectasis: Contraction Atelectasis (Or Cicatrization Atelectasis)Dokumen10 halamanAtelectasis: Contraction Atelectasis (Or Cicatrization Atelectasis)Gan BangBelum ada peringkat
- Management ARDS - Dr. AndriDokumen54 halamanManagement ARDS - Dr. AndrifandimuttaqinBelum ada peringkat
- Spirometry: How To Do The TestDokumen4 halamanSpirometry: How To Do The TestMarc AbellaBelum ada peringkat
- Module 1 - Oxygen Therapy (Student)Dokumen8 halamanModule 1 - Oxygen Therapy (Student)febie pachecoBelum ada peringkat
- Anesthesia MachineDokumen54 halamanAnesthesia MachineSador YonasBelum ada peringkat
- L5 - Control & Adjustment of RespirationDokumen24 halamanL5 - Control & Adjustment of Respirationzaini nieBelum ada peringkat
- Shock SepticDokumen35 halamanShock SepticAkbar SyarialBelum ada peringkat
- Mitochondria Is The Powerhouse of The CellDokumen2 halamanMitochondria Is The Powerhouse of The CellSenap AwitBelum ada peringkat