Anda mungkin juga menyukai
- Cycle de KrebsDokumen37 halamanCycle de KrebscepBelum ada peringkat
- Bêta OxydationDokumen7 halamanBêta OxydationAnass MaaroufiBelum ada peringkat
- Metabolisme Des Acides Gras 2018 PDFDokumen85 halamanMetabolisme Des Acides Gras 2018 PDFPace MakerBelum ada peringkat
- Biosynthése Des Lipidsynt10Dokumen9 halamanBiosynthése Des Lipidsynt10mhicheBelum ada peringkat
- CH3.Lipogénèse PFDokumen25 halamanCH3.Lipogénèse PFkhammar hichemBelum ada peringkat
- Métabolisme Des LipidesDokumen17 halamanMétabolisme Des LipidesMei Wan100% (1)
- Métabolisme Des LipidesDokumen47 halamanMétabolisme Des LipidesBoubaker AmiraBelum ada peringkat
- Glucides: Les Grands Articles d'UniversalisDari EverandGlucides: Les Grands Articles d'UniversalisBelum ada peringkat
- Metabolisme LipidesDokumen49 halamanMetabolisme LipidespartiraretirapasBelum ada peringkat
- Metabo Partie2Dokumen6 halamanMetabo Partie2la biochimiste100% (2)
- GlycolyseDokumen45 halamanGlycolysecepBelum ada peringkat
- Métabolisme Des Acides AminésDokumen40 halamanMétabolisme Des Acides AminéscepBelum ada peringkat
- Métabolisme Des LIPIDESDokumen9 halamanMétabolisme Des LIPIDESRahaf Fati100% (1)
- Métabolisme Des Glucides Fiche PDFDokumen7 halamanMétabolisme Des Glucides Fiche PDFNaïs LABBADIBelum ada peringkat
- Correction TD Metabolisme Glucidique 2Dokumen31 halamanCorrection TD Metabolisme Glucidique 2acheparfaite958Belum ada peringkat
- Metabolisme Des LipidesDokumen97 halamanMetabolisme Des LipidesFikra PapBelum ada peringkat
- Metabolisme Des Acides AminesDokumen12 halamanMetabolisme Des Acides AminesWissal ElbarBelum ada peringkat
- 2 - Le Métabolisme Des GlucidesDokumen12 halaman2 - Le Métabolisme Des GlucidesFanny Groux100% (2)
- NeoglucogeneseDokumen35 halamanNeoglucogenesecepBelum ada peringkat
- TD Biochimie+métabolique 2020 2021 DESKTOP 10K4H4KDokumen4 halamanTD Biochimie+métabolique 2020 2021 DESKTOP 10K4H4KAG PROMO MarocBelum ada peringkat
- La NéoglucogenèseDokumen34 halamanLa Néoglucogenèsevague2000Belum ada peringkat
- Peptides Et ProtéinesDokumen28 halamanPeptides Et ProtéinesFanny GrouxBelum ada peringkat
- TD Glucide Medecine 2020 2021 (Série)Dokumen4 halamanTD Glucide Medecine 2020 2021 (Série)Sarah BelbachirBelum ada peringkat
- LES LIPIDES Sructure Et Métabolque 2010Dokumen162 halamanLES LIPIDES Sructure Et Métabolque 2010Lyes DahmaniBelum ada peringkat
- Le Cycle de KrebsDokumen19 halamanLe Cycle de KrebsLandry RimbaybarBelum ada peringkat
- TD 3 Corrigé Regulation SABRINA.2019Dokumen5 halamanTD 3 Corrigé Regulation SABRINA.2019axelBelum ada peringkat
- Chapitre Voies MétaboliquesDokumen57 halamanChapitre Voies MétaboliquesMBAÏADOUM NGARGUINAM RodrigueBelum ada peringkat
- 5 Metabolisme 10 11Dokumen23 halaman5 Metabolisme 10 11Slip Horticole100% (1)
- MEtabolisme Des GlucidesDokumen16 halamanMEtabolisme Des GlucidesOtmane HajiBelum ada peringkat
- Metabolisme Des TriglycéridesDokumen24 halamanMetabolisme Des Triglycéridesvague2000Belum ada peringkat
- Sujet BiochimieDokumen5 halamanSujet BiochimieEmmanuella AssaBelum ada peringkat
- C. Biochimie III. Lipides (III.1. Métabolisme Des Acides Gras)Dokumen41 halamanC. Biochimie III. Lipides (III.1. Métabolisme Des Acides Gras)Rania HamdiBelum ada peringkat
- Correction Du TD de Biochimie 2021-2022Dokumen22 halamanCorrection Du TD de Biochimie 2021-2022Issouf Berthe100% (1)
- Métabolisme Des GlucidesDokumen6 halamanMétabolisme Des GlucidesHouda SoniaBelum ada peringkat
- TD Biochimie Métabolique 2022 2023Dokumen5 halamanTD Biochimie Métabolique 2022 2023AG PROMO MarocBelum ada peringkat
- CM Regulation Metabolique 2 Enzymes Et Allosterie 2019 ModDokumen31 halamanCM Regulation Metabolique 2 Enzymes Et Allosterie 2019 ModSirina LadjBelum ada peringkat
- Poly 03-Metabolisme Des GlucidesDokumen52 halamanPoly 03-Metabolisme Des Glucidessafemind100% (1)
- Métabolisme Des GlucidesDokumen28 halamanMétabolisme Des GlucidesilaBelum ada peringkat
- Metabolisme GlucidesDokumen20 halamanMetabolisme Glucidesscaren efonaBelum ada peringkat
- Les Acides AminesDokumen7 halamanLes Acides Aminesbouguidima_sanaBelum ada peringkat
- Metabolisme LipidesDokumen49 halamanMetabolisme LipidesMohamed Ali MtibaaBelum ada peringkat
- EMD Regroupés (Biochimie Glucides, Lipides Et AA)Dokumen11 halamanEMD Regroupés (Biochimie Glucides, Lipides Et AA)Hakim Rahmani0% (1)
- TD 2 PGF 2020-2021Dokumen2 halamanTD 2 PGF 2020-2021MounirBelum ada peringkat
- 12-Les CatecholaminesDokumen4 halaman12-Les CatecholaminesoutsiderBelum ada peringkat
- CHAP7 MET DES LIPIDES 1 Métabolisme Des Acides Gras 2022 2023 PLUSDokumen9 halamanCHAP7 MET DES LIPIDES 1 Métabolisme Des Acides Gras 2022 2023 PLUSdianaBelum ada peringkat
- Acides AminesDokumen20 halamanAcides Amines다리바야Belum ada peringkat
- Metabolisme Des Glucides PDFDokumen157 halamanMetabolisme Des Glucides PDFMariemBoujmal33% (3)
- C. Biochimie III. Lipides (I. Caractères Généraux Des Lipides)Dokumen68 halamanC. Biochimie III. Lipides (I. Caractères Généraux Des Lipides)Rania HamdiBelum ada peringkat
- Cahier Dexercices de Biochimie 5 MetabolismeDokumen15 halamanCahier Dexercices de Biochimie 5 Metabolismeيحيى بورغدةBelum ada peringkat
- TD (Métabolisme Des Glucides)Dokumen1 halamanTD (Métabolisme Des Glucides)يحيى بورغدة100% (1)
- Cycle de Krebs - Biochimie Métabolique - Hader HaidousDokumen13 halamanCycle de Krebs - Biochimie Métabolique - Hader Haidousrobbihad100% (3)
- 02 Structure Cyclique Des OsesDokumen10 halaman02 Structure Cyclique Des OsesBook Hunter100% (1)
- TD Enzymo Biochimie Met 1Dokumen31 halamanTD Enzymo Biochimie Met 1Saminat RidjaliBelum ada peringkat
- NeoglucogeneseDokumen29 halamanNeoglucogeneseHassan ElabdiBelum ada peringkat
- TD Acides Aminés Et PeptidesDokumen2 halamanTD Acides Aminés Et PeptidesNaruto & SasukeBelum ada peringkat
- Biochimie2an-Equilibre Acidobasique2021demmakDokumen15 halamanBiochimie2an-Equilibre Acidobasique2021demmakGlobule RougeBelum ada peringkat
- Applications de la spectrophotomérie en phytochimie: sciencesDari EverandApplications de la spectrophotomérie en phytochimie: sciencesBelum ada peringkat
- Acides nucléiques: Les Grands Articles d'UniversalisDari EverandAcides nucléiques: Les Grands Articles d'UniversalisBelum ada peringkat
- 002 - Cours GlycolyseDokumen25 halaman002 - Cours GlycolyseBlueskyindanBelum ada peringkat
- 002 - Cours Métabolisme Du Cholestérol PDFDokumen8 halaman002 - Cours Métabolisme Du Cholestérol PDFBlueskyindanBelum ada peringkat
- 001 - Cours NéoglucogenèseDokumen13 halaman001 - Cours NéoglucogenèseBlueskyindanBelum ada peringkat
- 003 - Cours Métabolisme Des Corps Cétoniques PDFDokumen5 halaman003 - Cours Métabolisme Des Corps Cétoniques PDFBlueskyindan100% (1)
- 003 - Régulation de L - Activité EnzymatiqueDokumen43 halaman003 - Régulation de L - Activité EnzymatiqueBlueskyindanBelum ada peringkat
- 003 - Régulation de L - Activité EnzymatiqueDokumen43 halaman003 - Régulation de L - Activité EnzymatiqueBlueskyindanBelum ada peringkat
- 003 - Régulation de L - Activité EnzymatiqueDokumen43 halaman003 - Régulation de L - Activité EnzymatiqueBlueskyindanBelum ada peringkat
- 003 - Régulation de L - Activité EnzymatiqueDokumen15 halaman003 - Régulation de L - Activité EnzymatiqueBlueskyindanBelum ada peringkat
- 003 - Régulation de L - Activité EnzymatiqueDokumen15 halaman003 - Régulation de L - Activité EnzymatiqueBlueskyindanBelum ada peringkat
- 003 - Régulation de L - Activité EnzymatiqueDokumen15 halaman003 - Régulation de L - Activité EnzymatiqueBlueskyindanBelum ada peringkat
- 003 - Régulation de L - Activité EnzymatiqueDokumen15 halaman003 - Régulation de L - Activité EnzymatiqueBlueskyindanBelum ada peringkat
- 003 - Régulation de L - Activité EnzymatiqueDokumen15 halaman003 - Régulation de L - Activité EnzymatiqueBlueskyindanBelum ada peringkat
- Devoir de Première S N°2 - Type II2Dokumen3 halamanDevoir de Première S N°2 - Type II2Nihed BelhadjiBelum ada peringkat
- 21 Dermatoses Bulleuses - Cas CliniquesDokumen208 halaman21 Dermatoses Bulleuses - Cas CliniquesKada Ben youcefBelum ada peringkat
- Cours N1 Mecanisme D - Action Des Xenobiotiques 2016Dokumen23 halamanCours N1 Mecanisme D - Action Des Xenobiotiques 2016souane ibrahim25% (4)
- FécondationDokumen10 halamanFécondationhauswirthBelum ada peringkat
- Klein 2014Dokumen17 halamanKlein 2014ImaneBelum ada peringkat
- ImmunologieDokumen6 halamanImmunologiemalak wxBelum ada peringkat
- Cours de Biochimie Métabolisme Phosphocalcique Première Année Médecine DentaireDokumen85 halamanCours de Biochimie Métabolisme Phosphocalcique Première Année Médecine DentaireNadar ArbiBelum ada peringkat
- Thyroide (Dr. Bouazdi 2016)Dokumen68 halamanThyroide (Dr. Bouazdi 2016)Dounya BenyellesBelum ada peringkat
- Chapitre 2 Tissu Conjonctif LAMDA 2021Dokumen40 halamanChapitre 2 Tissu Conjonctif LAMDA 2021LàkàmoràEnAlgérieBelum ada peringkat
- Techniques D'immobilisation Des Cellules Et Leur ApplicationDokumen50 halamanTechniques D'immobilisation Des Cellules Et Leur Applicationhouhou_houhou83% (24)
- Acide NucleiqueDokumen4 halamanAcide NucleiqueAli GohBelum ada peringkat
- Capacité D'adaptation Au Changement Climatique À MadagascarDokumen2 halamanCapacité D'adaptation Au Changement Climatique À MadagascarHayZara Madagascar100% (1)
- Niveau X ClassificationsDokumen4 halamanNiveau X ClassificationsMOZARGODWINBelum ada peringkat
- 02 La Guilde Du CerisierDokumen9 halaman02 La Guilde Du CerisierHerbe De LuneBelum ada peringkat
- Les Quinze Premières Vies DHarry August (North Claire (North Claire) ) (Z-Library)Dokumen404 halamanLes Quinze Premières Vies DHarry August (North Claire (North Claire) ) (Z-Library)Cécile AlexandriaBelum ada peringkat
- Evaluation Os MusclesDokumen2 halamanEvaluation Os MusclesNiangBelum ada peringkat
- HACHANADokumen10 halamanHACHANALyna BkrBelum ada peringkat
- Cours de Biologie Les EnzymesDokumen10 halamanCours de Biologie Les EnzymesOussamaBelum ada peringkat
- (CCS) (2003) - ChimieDokumen9 halaman(CCS) (2003) - ChimiebabibenBelum ada peringkat
- Null PDFDokumen26 halamanNull PDFAziz OualidBelum ada peringkat
- Le Palmier A HuileDokumen6 halamanLe Palmier A HuileFrogy SebastianBelum ada peringkat
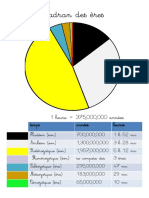
- Cadran Des ÈresDokumen11 halamanCadran Des ÈresFrancofolieBelum ada peringkat
- Commentaire LectriceDokumen3 halamanCommentaire LectriceSiham BouhaBelum ada peringkat
- Cardio HallaDokumen53 halamanCardio HallaTewfik TGrBelum ada peringkat
- PE 13 14 PrepaDokumen12 halamanPE 13 14 Prepamahmoud sfar hanchaBelum ada peringkat
- 572 01088 PDFDokumen79 halaman572 01088 PDFmaroua henkaBelum ada peringkat
- Cours 5 PDFDokumen47 halamanCours 5 PDFÑã DåBelum ada peringkat
- Alimentation Et État Nutritionnel Des Enfants de 0-36 Mois Dans La Commune Rurale de Sabotsy Namehana (MAHAZAFY - INSPC/2007)Dokumen61 halamanAlimentation Et État Nutritionnel Des Enfants de 0-36 Mois Dans La Commune Rurale de Sabotsy Namehana (MAHAZAFY - INSPC/2007)HayZara MadagascarBelum ada peringkat
- Les Plantes Utiles Du Gabon Journal D'agriculture Tropicale Etde Botanique AppliquéeDokumen32 halamanLes Plantes Utiles Du Gabon Journal D'agriculture Tropicale Etde Botanique AppliquéekalyBelum ada peringkat
- Eckert - Fisiologia Animal PDFDokumen621 halamanEckert - Fisiologia Animal PDFJesús LabGomBelum ada peringkat