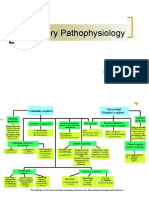
Anda mungkin juga menyukai
- Pulmonary ConditionsDokumen42 halamanPulmonary ConditionsMinetteBelum ada peringkat
- The Best Way To Make Your Dreams Come True Is To: Wake Up..Dokumen28 halamanThe Best Way To Make Your Dreams Come True Is To: Wake Up..Chandan Kumar SinghBelum ada peringkat
- Suppurative Lung DiseaseDokumen45 halamanSuppurative Lung DiseasemusabBelum ada peringkat
- Respiratory Pathology: Dr. Okon MRCSDokumen38 halamanRespiratory Pathology: Dr. Okon MRCSEdwin OkonBelum ada peringkat
- Pathology Slide #6+7Dokumen73 halamanPathology Slide #6+7Ibrahim MigdadyBelum ada peringkat
- Topic 5 Lobar PneumoniaDokumen16 halamanTopic 5 Lobar PneumoniaAmro AzrakBelum ada peringkat
- Week 4 Restrictive DisordersDokumen212 halamanWeek 4 Restrictive Disordersdelrosariojm87Belum ada peringkat
- Respiratory Pathophysiology: B. Pimentel, M.D. University of Makati College of NursingDokumen12 halamanRespiratory Pathophysiology: B. Pimentel, M.D. University of Makati College of NursingDoc JacqueBelum ada peringkat
- Clinical Physiology of Respiration: Dr. M Qathar RF TDokumen76 halamanClinical Physiology of Respiration: Dr. M Qathar RF TTiwi Lestari TiwiBelum ada peringkat
- Respiratory: Welcome!Dokumen119 halamanRespiratory: Welcome!Majo ParagasBelum ada peringkat
- Pathology of Airway DiseasesDokumen42 halamanPathology of Airway DiseasesZijieBelum ada peringkat
- Respiration CH 43.Dr SarahDokumen59 halamanRespiration CH 43.Dr Sarahaiman siddiquiBelum ada peringkat
- EmpyemaDokumen52 halamanEmpyemaDivara Syauta100% (3)
- 6.lung AbscessDokumen13 halaman6.lung AbscessManushi HenadeeraBelum ada peringkat
- CoughDokumen30 halamanCoughAnil NarayanBelum ada peringkat
- Bronchial ObstructionDokumen137 halamanBronchial ObstructionGrajdianu Natalia100% (1)
- Bronchiectasis OkDokumen60 halamanBronchiectasis OkImmanuelBelum ada peringkat
- Respiratory Failure: by Amera GumamaDokumen26 halamanRespiratory Failure: by Amera GumamaGumama AmeiyrhaBelum ada peringkat
- Respiratory Disorders in ChildrenDokumen77 halamanRespiratory Disorders in ChildrenJharaBelum ada peringkat
- Suppurative Lung Diseases: DR Taher El Naggar Prof of Pulmonary Medicine Ain Shams UniversityDokumen65 halamanSuppurative Lung Diseases: DR Taher El Naggar Prof of Pulmonary Medicine Ain Shams UniversitykingmedicBelum ada peringkat
- Lung AbscessDokumen38 halamanLung Abscessprabad dunusingheBelum ada peringkat
- Lung Abscess: Presented byDokumen36 halamanLung Abscess: Presented byPalanki Gopal100% (1)
- Wk4 Restrictive Lung DisordersDokumen38 halamanWk4 Restrictive Lung DisordersPotato PceeBelum ada peringkat
- PneumoniaDokumen24 halamanPneumoniaMuhammad HassanBelum ada peringkat
- Approach To A Child With Cough and Difficulty in BreathingDokumen23 halamanApproach To A Child With Cough and Difficulty in BreathingKashif Burki100% (2)
- B&lung AbscessDokumen21 halamanB&lung Abscessnathan asfahaBelum ada peringkat
- Lung AbscessDokumen19 halamanLung Abscessvtyagi2377Belum ada peringkat
- Broncho PneumoniaDokumen21 halamanBroncho PneumoniaRiad GagahBelum ada peringkat
- BRONCHIECTASISDokumen40 halamanBRONCHIECTASISImmanuel100% (2)
- Lower Respiratory Disorders Part 1Dokumen70 halamanLower Respiratory Disorders Part 1Joseph Krafft100% (1)
- Lung AbscessDokumen41 halamanLung AbscessokaciaBelum ada peringkat
- AtelectasisDokumen26 halamanAtelectasisArini NurlelaBelum ada peringkat
- PneumothoraxDokumen19 halamanPneumothoraxMonika StephyBelum ada peringkat
- Pneumonia ConsultationDokumen64 halamanPneumonia ConsultationYustika PutriBelum ada peringkat
- Pulmonary PathologyDokumen46 halamanPulmonary PathologyMuostafa KamelBelum ada peringkat
- Tension Pneumothorax by AnkurDokumen30 halamanTension Pneumothorax by AnkurAnkur AgrawalBelum ada peringkat
- Sistem Respirasi Sesak Napas: Problem Based LearningDokumen61 halamanSistem Respirasi Sesak Napas: Problem Based LearningAkbar IskandarBelum ada peringkat
- Notes SC RSDokumen5 halamanNotes SC RS202213Belum ada peringkat
- Lung AbcessDokumen12 halamanLung AbcessABI OFFICIALBelum ada peringkat
- Part III Internal Medicine Examination AnswersDokumen91 halamanPart III Internal Medicine Examination AnswersFırat GüllüBelum ada peringkat
- Respiratory System SummaryDokumen6 halamanRespiratory System SummaryKiara GovenderBelum ada peringkat
- PneumothoraxDokumen43 halamanPneumothoraxSravani KanchiBelum ada peringkat
- Abses ParuDokumen16 halamanAbses ParuNadiya SafitriBelum ada peringkat
- Bronchiectasis: Dr. Kapil Rastogi MPT (Cardiopulmonary) Assistant ProfessorDokumen18 halamanBronchiectasis: Dr. Kapil Rastogi MPT (Cardiopulmonary) Assistant ProfessorKavya MittalBelum ada peringkat
- Pneumothorax and Pneumomediastinum: Dr. Emad EfatDokumen89 halamanPneumothorax and Pneumomediastinum: Dr. Emad Efatinterna MANADOBelum ada peringkat
- PATHOPHYSIOLOGYDokumen46 halamanPATHOPHYSIOLOGYnimrakhalid82000Belum ada peringkat
- Professor Youry Vladimirovitch PlotnicovDokumen109 halamanProfessor Youry Vladimirovitch PlotnicovOxana TulbeaBelum ada peringkat
- 1respiratory System DisordersDokumen10 halaman1respiratory System DisordersArvin MalondrasBelum ada peringkat
- 2 Mark Questions and Answers For Croup Syndrome, Cystic Fibrosis, Bronchitis, Bronchiolitis and Bronchiectasis Define Croup SyndromeDokumen5 halaman2 Mark Questions and Answers For Croup Syndrome, Cystic Fibrosis, Bronchitis, Bronchiolitis and Bronchiectasis Define Croup SyndromeVignesh KumarBelum ada peringkat
- Study Notes Respiratory SystemDokumen19 halamanStudy Notes Respiratory SystemAnde Mangkuluhur Azhari ThalibbanBelum ada peringkat
- Acute Lung Abscesses. Definition of The Idea. Classification. Etiology and Pathogenesis. Clinical Picture. Diagnosis.Dokumen6 halamanAcute Lung Abscesses. Definition of The Idea. Classification. Etiology and Pathogenesis. Clinical Picture. Diagnosis.Lucas Victor AlmeidaBelum ada peringkat
- MSNDokumen16 halamanMSNVikram bhai prajapatiBelum ada peringkat
- Pulmonary Examination - Knowledge at AMBOSSDokumen1 halamanPulmonary Examination - Knowledge at AMBOSSKC Dela RosaBelum ada peringkat
- Respiratorydisease 170426125838Dokumen69 halamanRespiratorydisease 170426125838Hayder MaqsadBelum ada peringkat
- Mr. Maheboob 1 Year M.SC Nursing Govt College of Nursing HolenarsipurDokumen44 halamanMr. Maheboob 1 Year M.SC Nursing Govt College of Nursing HolenarsipurDr Tahira NihalBelum ada peringkat
- Lung Reviewer PathologyDokumen9 halamanLung Reviewer PathologyTee VillanuevaBelum ada peringkat
- PneumoniaDokumen3 halamanPneumoniaKami KhanBelum ada peringkat
- W1 L2 PneumoniaDokumen57 halamanW1 L2 PneumoniaAnas FikriBelum ada peringkat
- Pleural EffusionDokumen24 halamanPleural Effusionwheeyycoldandhot55Belum ada peringkat
- Medical Mnemonic Sketches : Pulmonary DiseasesDari EverandMedical Mnemonic Sketches : Pulmonary DiseasesBelum ada peringkat
- TrematodesDokumen1 halamanTrematodesfatima chrystelle nuñalBelum ada peringkat
- Postoperative Ileus: Cristina R. Harnsberger, MD Justin A. Maykel, MD Karim Alavi, MD, MPHDokumen5 halamanPostoperative Ileus: Cristina R. Harnsberger, MD Justin A. Maykel, MD Karim Alavi, MD, MPHfatima chrystelle nuñalBelum ada peringkat
- Urological Injuries During Colorectal SurgeryDokumen8 halamanUrological Injuries During Colorectal Surgeryfatima chrystelle nuñalBelum ada peringkat
- Community Acquired Pneumonia Nelson 400Dokumen37 halamanCommunity Acquired Pneumonia Nelson 400fatima chrystelle nuñalBelum ada peringkat
- Acute GastroenteritisDokumen31 halamanAcute Gastroenteritisfatima chrystelle nuñalBelum ada peringkat
- MeckelDokumen6 halamanMeckelfatima chrystelle nuñalBelum ada peringkat
- Essential Intrapartum Newborn CareDokumen36 halamanEssential Intrapartum Newborn Carefatima chrystelle nuñal100% (1)
- Breast QDokumen6 halamanBreast Qfatima chrystelle nuñalBelum ada peringkat
- Anesthesia MCQDokumen5 halamanAnesthesia MCQfatima chrystelle nuñal100% (1)
- What Are Thyroid DisordersDokumen1 halamanWhat Are Thyroid Disordersfatima chrystelle nuñalBelum ada peringkat
- Breast QDokumen6 halamanBreast Qfatima chrystelle nuñalBelum ada peringkat
- Assessment of The Respiratory SystemDokumen49 halamanAssessment of The Respiratory SystemMariana Mikaela AlagarBelum ada peringkat
- Positive Expiratory Pressure and PDFDokumen20 halamanPositive Expiratory Pressure and PDFPaoly Palma100% (2)
- 10 PulmunologyDokumen111 halaman10 PulmunologyJeet JainBelum ada peringkat
- 2 - Respiratory Notes MRCP-I Passmedicine 2017Dokumen92 halaman2 - Respiratory Notes MRCP-I Passmedicine 2017kathi raja sekhar100% (1)
- Autogenic Drainage Exercises: Community Respiratory ServiceDokumen3 halamanAutogenic Drainage Exercises: Community Respiratory ServiceKaren BaldionBelum ada peringkat
- Airway-Centered Fibroelastosis 2016Dokumen8 halamanAirway-Centered Fibroelastosis 2016Edoardo CavigliBelum ada peringkat
- Differential Diagnosis of Acute Exacerbation of CopdDokumen4 halamanDifferential Diagnosis of Acute Exacerbation of CopdManik ParmeliaBelum ada peringkat
- ICD Reference CodesDokumen2 halamanICD Reference CodesRavi Pal100% (1)
- Respiratory Questions by DR - OkashaDokumen96 halamanRespiratory Questions by DR - Okashamohammed okashaBelum ada peringkat
- Surgical Diseases. Radio-Imagistic Investigations: USMF "N.Testemitanu"Dokumen156 halamanSurgical Diseases. Radio-Imagistic Investigations: USMF "N.Testemitanu"andrewBelum ada peringkat
- Allergic Bronchopulmonary Aspergillasis: The Spectrum of Roentgenologic AppearancesDokumen31 halamanAllergic Bronchopulmonary Aspergillasis: The Spectrum of Roentgenologic Appearancesmahak chaudharyBelum ada peringkat
- Respiratory Pastest PDFDokumen711 halamanRespiratory Pastest PDFDr-Jahanzaib GondalBelum ada peringkat
- Chapter 012 QuestionsDokumen35 halamanChapter 012 QuestionsPrecilou CutandaBelum ada peringkat
- Long Case Mmbs 06 11Dokumen110 halamanLong Case Mmbs 06 11whee182Belum ada peringkat
- Asthma: Chronic Inflammation of The Airways. The Hyper-Responsive Airways Typical of AsthmaDokumen7 halamanAsthma: Chronic Inflammation of The Airways. The Hyper-Responsive Airways Typical of AsthmaRaj PaulBelum ada peringkat
- Bronchiectasis: PathophysiologyDokumen3 halamanBronchiectasis: PathophysiologyBharat Singh BanshiwalBelum ada peringkat
- Respiratory Symptoms and Signs: Key PointsDokumen9 halamanRespiratory Symptoms and Signs: Key PointsAna LopezBelum ada peringkat
- Gerontological Nursing 2021Dokumen157 halamanGerontological Nursing 2021Andrew Isiah BonifacioBelum ada peringkat
- Pulmonary and Critical Care MnemonicsDokumen10 halamanPulmonary and Critical Care MnemonicsnmahpbooksBelum ada peringkat
- Suppurative Lung Diseases..Dokumen61 halamanSuppurative Lung Diseases..Salman KhanBelum ada peringkat
- Respiratory Passmedicine 2020Dokumen555 halamanRespiratory Passmedicine 2020Vikrant100% (2)
- NCPDokumen3 halamanNCPNikki del Rosario100% (2)
- Suppurative Desease of Lungs and PleuraDokumen26 halamanSuppurative Desease of Lungs and PleuraGarima SahuBelum ada peringkat
- Cardio Pulmo NotesDokumen12 halamanCardio Pulmo NotesCherrie MaeBelum ada peringkat
- Community-Acquired Pneumoniaandhospital-Acquiredpneumonia: Charles W. Lanks,, Ali I. Musani,, David W. HsiaDokumen15 halamanCommunity-Acquired Pneumoniaandhospital-Acquiredpneumonia: Charles W. Lanks,, Ali I. Musani,, David W. HsiaMajo EscobarBelum ada peringkat
- Bronchiectasis: Case & ReviewDokumen40 halamanBronchiectasis: Case & ReviewKhaled S. HarbBelum ada peringkat
- Core Clinical ProblemsDokumen57 halamanCore Clinical ProblemsPatriciaChRistianiBelum ada peringkat
- The Future of Cystic Fibrosis Treatment From DiseDokumen14 halamanThe Future of Cystic Fibrosis Treatment From DiseAlexander LozaBelum ada peringkat
- Respiratory History and ExaminationDokumen7 halamanRespiratory History and Examinationjen262004Belum ada peringkat
- Pidotimod In-Depth Review of Current EvidenceDokumen12 halamanPidotimod In-Depth Review of Current EvidenceColider ISHMEDBelum ada peringkat