Anda mungkin juga menyukai
- UrosepsisDokumen7 halamanUrosepsisOrlan PeBoBelum ada peringkat
- Atencion de Enfermeria en El Triage 2019Dokumen150 halamanAtencion de Enfermeria en El Triage 2019Stella100% (2)
- Regulación del volumen extracelular y rol del riñónDokumen7 halamanRegulación del volumen extracelular y rol del riñónMarlenAlcantaraSerranoBelum ada peringkat
- Escalas en EnfermeriaDokumen9 halamanEscalas en EnfermeriaJudith Asuncion Lopez Sanchez100% (3)
- Farmacologia Renal PDFDokumen13 halamanFarmacologia Renal PDFVera ItzelBelum ada peringkat
- AlbuminaDokumen3 halamanAlbuminaLeticia EstefaniaBelum ada peringkat
- Química Clase 4Dokumen5 halamanQuímica Clase 4jose bBelum ada peringkat
- 01 Introducción Fisica MariscalDokumen26 halaman01 Introducción Fisica MariscalAndy LilaBelum ada peringkat
- Cap 23 GeometriaDokumen6 halamanCap 23 Geometriaerwin_carryBelum ada peringkat
- Tech Manual-SPDokumen27 halamanTech Manual-SPJoaquínIgnacioBelum ada peringkat
- Historias Con Perros y GatosDokumen15 halamanHistorias Con Perros y GatosSoledad ZanninoBelum ada peringkat
- Evaluacion Del Cuidado de ManosDokumen5 halamanEvaluacion Del Cuidado de ManosAlberta Martinez Gomez100% (1)
- ¿Niño o Niña El Ser Humano Posee 46 Cromosomas en Cada CelulabDokumen162 halaman¿Niño o Niña El Ser Humano Posee 46 Cromosomas en Cada Celulabe5490626Belum ada peringkat
- Mapa Mental Lean ManufacturingDokumen4 halamanMapa Mental Lean ManufacturingMemo Gonzalez Aleman100% (2)
- Monografía Latin 2Dokumen11 halamanMonografía Latin 2Gabriel Andres RaveraBelum ada peringkat
- Ic303 T7 Techint Reporte - FinalDokumen7 halamanIc303 T7 Techint Reporte - FinalDaniel CutimboBelum ada peringkat
- 4 Memoria de Internado de Enfermeria 2020Dokumen112 halaman4 Memoria de Internado de Enfermeria 2020Ketty Laura VilcapomaBelum ada peringkat
- Nueva Calle: Plano Topografico de Campo Deportivo Recreacional - Acobamba-HvcaDokumen1 halamanNueva Calle: Plano Topografico de Campo Deportivo Recreacional - Acobamba-HvcaLina ApumaytaBelum ada peringkat
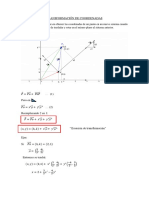
- Transformación de CoordenadasDokumen3 halamanTransformación de CoordenadasJimy el rosqueteBelum ada peringkat
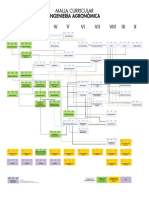
- Malla Curricular Ing Agronómica UNALDokumen1 halamanMalla Curricular Ing Agronómica UNALkmigeBelum ada peringkat
- CancioneroDokumen5 halamanCancioneroNatalí MirandaBelum ada peringkat
- Hoja de Seguridad Pinturas A Base de AguaDokumen5 halamanHoja de Seguridad Pinturas A Base de AguaNESTOR RAUL BERGANO HERRERA100% (1)
- Adivinanzas - para Niños Con Respuestas. 12 - 10 - 10Dokumen3 halamanAdivinanzas - para Niños Con Respuestas. 12 - 10 - 10LIPANURUBelum ada peringkat
- Práctica 1. Precisión y Exactitud.Dokumen7 halamanPráctica 1. Precisión y Exactitud.Ulises MolinaBelum ada peringkat
- El Hombre Ser EducableDokumen30 halamanEl Hombre Ser EducableLicenciatura HistoriaBelum ada peringkat
- Ciclo de KrebsDokumen9 halamanCiclo de Krebsapi-235957287Belum ada peringkat
- Neoplasia SDokumen16 halamanNeoplasia SSG CarolinaBelum ada peringkat
- Gestion de Compras e InventariosDokumen26 halamanGestion de Compras e Inventariosalexander peña jBelum ada peringkat
- Lab Teorema de THEVENIN y NORTON en Regimen ACDokumen6 halamanLab Teorema de THEVENIN y NORTON en Regimen ACLEONARDO BOLIVAR VILLEGASBelum ada peringkat
- Ingeniería Mecánica y su Impacto en la SociedadDokumen11 halamanIngeniería Mecánica y su Impacto en la SociedadAlejandro ValeroBelum ada peringkat
- Trabajo Fisica 1 Lab 2Dokumen37 halamanTrabajo Fisica 1 Lab 2Alexander KuroSake GuevaraBelum ada peringkat
- 2016-03 Top Value Computing v1 PDFDokumen24 halaman2016-03 Top Value Computing v1 PDFJaime TraverBelum ada peringkat
- Sec PD ArtesVisuales3 AG-1Dokumen84 halamanSec PD ArtesVisuales3 AG-1Lupita Glez80% (5)
- PDF - Master Unidad 2Dokumen183 halamanPDF - Master Unidad 2Gustavo Diego TGBelum ada peringkat
- Diseño de mezcla de concreto hidráulico para 270 kg/cm2Dokumen11 halamanDiseño de mezcla de concreto hidráulico para 270 kg/cm2Nitram Lopez TlehuactleBelum ada peringkat
- Secretos Del Perfecto Actor de TeatroDokumen46 halamanSecretos Del Perfecto Actor de TeatroJosue Manuel Rodriguez del ValleBelum ada peringkat