Anda mungkin juga menyukai
- Sindrome de RapunzelDokumen3 halamanSindrome de Rapunzelstefany alvarezBelum ada peringkat
- Indicaciones Contraindicaciones y Deficit de EstrogenosDokumen5 halamanIndicaciones Contraindicaciones y Deficit de EstrogenosThalía Lelis Sánchez SantillánBelum ada peringkat
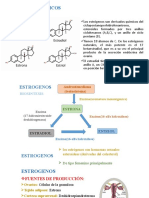
- Estrogenos, Estructura QuimicaDokumen13 halamanEstrogenos, Estructura QuimicaThalía Lelis Sánchez Santillán0% (1)
- Ponentes AntibioticosDokumen3 halamanPonentes AntibioticosThalía Lelis Sánchez SantillánBelum ada peringkat
- Dolor GeneralDokumen2 halamanDolor GeneralThalía Lelis Sánchez SantillánBelum ada peringkat
- Seram2014 S-0806Dokumen32 halamanSeram2014 S-0806Thalía Lelis Sánchez SantillánBelum ada peringkat
- TROMBOCITOSISDokumen19 halamanTROMBOCITOSISThalía Lelis Sánchez Santillán50% (2)
- ResistenciaDokumen18 halamanResistenciaThalía Lelis Sánchez SantillánBelum ada peringkat
- SalmeterolDokumen9 halamanSalmeterolThalía Lelis Sánchez SantillánBelum ada peringkat
- Como Ayudar Desde La Escuela A Un Niño Con TdahDokumen2 halamanComo Ayudar Desde La Escuela A Un Niño Con TdahanaliaBelum ada peringkat
- Examen Parcial I - S.IDokumen6 halamanExamen Parcial I - S.IDenis RodriguezBelum ada peringkat
- FO-050-11 (22-10-22) Incidente VV-101Dokumen3 halamanFO-050-11 (22-10-22) Incidente VV-101Diego Rodriguez GuechaBelum ada peringkat
- Situación de Violencia Contra Mujeres en ApurímacDokumen20 halamanSituación de Violencia Contra Mujeres en ApurímacSandra CajasBelum ada peringkat
- Ut 5 La MotivacionDokumen15 halamanUt 5 La Motivacionmaria_1960Belum ada peringkat
- Maestro Taller 2A. Conocimiento de La Norma ISO 22000Dokumen3 halamanMaestro Taller 2A. Conocimiento de La Norma ISO 22000brendacamachoBelum ada peringkat
- Trabajo de Investigacion de Proyecto Final Cuarto CapituloDokumen45 halamanTrabajo de Investigacion de Proyecto Final Cuarto CapituloItzel LopezBelum ada peringkat
- Intervencion Psicologica Bloque PsicosocialDokumen8 halamanIntervencion Psicologica Bloque PsicosocialMarta RoigBelum ada peringkat
- Enfe Unidad 6 (7) Digestivo 2023 PDFDokumen24 halamanEnfe Unidad 6 (7) Digestivo 2023 PDFJONATAN LUIS LEDESMABelum ada peringkat
- Orden No 153299-908000Dokumen1 halamanOrden No 153299-908000Laura Cerpa BaezBelum ada peringkat
- Indicación y Espacio para Enviar Tarea DE PSICOLOGIA Casi TerminadaDokumen8 halamanIndicación y Espacio para Enviar Tarea DE PSICOLOGIA Casi TerminadaMariaBelum ada peringkat
- Anexo B Formato de Inspección Centrales de GasesDokumen9 halamanAnexo B Formato de Inspección Centrales de GasesPaola del Rocio Garcia NavarreteBelum ada peringkat
- ReparaciónDokumen31 halamanReparaciónÁlvaro GarciaBelum ada peringkat
- Taller de OstomiasDokumen46 halamanTaller de OstomiasLUZ ANTONIA BARRETO ESPINOZABelum ada peringkat
- Ejemplos de Acreditamiento de IVADokumen13 halamanEjemplos de Acreditamiento de IVAAlejandra UnzuetaBelum ada peringkat
- Prac 03 BacterDokumen7 halamanPrac 03 BacterCinthia AlvarezBelum ada peringkat
- Percepción, Atribución, Actitud y DecisiónDokumen15 halamanPercepción, Atribución, Actitud y DecisiónAna Bella Castro GavidiaBelum ada peringkat
- Glosario TerminosDokumen4 halamanGlosario Terminosluis ospinoBelum ada peringkat
- Solicitud de EmpleoDokumen1 halamanSolicitud de EmpleoChristopher L'cBelum ada peringkat
- ASERTIVIDADDokumen11 halamanASERTIVIDADFernando AlabarBelum ada peringkat
- Orientaciones CLEI IVDokumen2 halamanOrientaciones CLEI IVCarlotaMonttBelum ada peringkat
- EHD Miller Developing FetalDokumen27 halamanEHD Miller Developing FetalCamilaBelum ada peringkat
- Regulación de Medicamentos Herbolarios y RemediosDokumen1 halamanRegulación de Medicamentos Herbolarios y RemediosmmarlenemeloBelum ada peringkat
- Tejido Psíquico Infanto-Juvenil (Parte 2)Dokumen15 halamanTejido Psíquico Infanto-Juvenil (Parte 2)Barbara QuetglasBelum ada peringkat
- Definición de EpidemiologíaDokumen1 halamanDefinición de EpidemiologíaBlanca Edith Agredano DuránBelum ada peringkat
- Revista MD PasionAnimal 5Dokumen40 halamanRevista MD PasionAnimal 5Ramsey MoralesBelum ada peringkat
- Acute Placental Abruption - Pathophysiology, Clinical Features, Diagnosis, and Consequences - UpToDateDokumen3 halamanAcute Placental Abruption - Pathophysiology, Clinical Features, Diagnosis, and Consequences - UpToDategerardo castellanosBelum ada peringkat
- Tarea - Semana 2 - LUIS SALLERESDokumen5 halamanTarea - Semana 2 - LUIS SALLERESLuis SalleresBelum ada peringkat
- Caso No Hay Que Olvidar Los PrincipiosDokumen2 halamanCaso No Hay Que Olvidar Los Principioselisban martinBelum ada peringkat