Anda mungkin juga menyukai
- Mathematical Foundations of Information TheoryDari EverandMathematical Foundations of Information TheoryPenilaian: 3.5 dari 5 bintang3.5/5 (9)
- A More Stable Approach To LISP Tree GP: 1 BackgroundDokumen11 halamanA More Stable Approach To LISP Tree GP: 1 BackgroundEpic WinBelum ada peringkat
- Dancing LinksDokumen26 halamanDancing LinksVegwynBelum ada peringkat
- Advanced Algorithms Course. Lecture Notes. Part 11: Chernoff BoundsDokumen4 halamanAdvanced Algorithms Course. Lecture Notes. Part 11: Chernoff BoundsKasapaBelum ada peringkat
- Topic 11: Autocorrelation: ECO2009: Empirical Economic AnalysisDokumen27 halamanTopic 11: Autocorrelation: ECO2009: Empirical Economic Analysissmurphy1234Belum ada peringkat
- David F. Shanno Roman L. Weil: Operations Research, Vol. 19, No. 1. (Jan. - Feb., 1971), Pp. 120-124Dokumen7 halamanDavid F. Shanno Roman L. Weil: Operations Research, Vol. 19, No. 1. (Jan. - Feb., 1971), Pp. 120-124Cơ Đinh VănBelum ada peringkat
- Calculus Non-Routine Problems and SolutionsDokumen51 halamanCalculus Non-Routine Problems and Solutionskaushik247Belum ada peringkat
- Regression Analysis - STAT510Dokumen34 halamanRegression Analysis - STAT510Vivian TranBelum ada peringkat
- SH Ann OnDokumen11 halamanSH Ann OnwaltzaBelum ada peringkat
- Operations ResearchDokumen37 halamanOperations ResearchShashank BadkulBelum ada peringkat
- On The Complexity of Protein FoldingDokumen36 halamanOn The Complexity of Protein Foldingeleni23Belum ada peringkat
- Numerical Solution To The Laplace EquationDokumen8 halamanNumerical Solution To The Laplace EquationabimanaBelum ada peringkat
- The Pharmacokinetic and Pharmacodynamic Basis of Target Controlled InfusionDokumen62 halamanThe Pharmacokinetic and Pharmacodynamic Basis of Target Controlled InfusioncaterinaBelum ada peringkat
- Project 1 (1) 1Dokumen24 halamanProject 1 (1) 121UMAR014 Arul Raj. SBelum ada peringkat
- VI. Notes (Played) On The Vibrating StringDokumen20 halamanVI. Notes (Played) On The Vibrating Stringjose2017Belum ada peringkat
- Monte Carlo Simulation LabDokumen9 halamanMonte Carlo Simulation LabS DasguptaBelum ada peringkat
- ANU MATH2405 Notes - 1Dokumen40 halamanANU MATH2405 Notes - 1James KingBelum ada peringkat
- 91850v00 NN10 CleveDokumen4 halaman91850v00 NN10 CleveVijay KumarBelum ada peringkat
- Electricity and Magnetism Online Laboratory Manual: Gavin A. BuxtonDokumen30 halamanElectricity and Magnetism Online Laboratory Manual: Gavin A. BuxtonUmair AbbasBelum ada peringkat
- Inverse Iteration Method For Finding EigenvectorsDokumen4 halamanInverse Iteration Method For Finding Eigenvectors王轩Belum ada peringkat
- Baum Chapter10 PDFDokumen29 halamanBaum Chapter10 PDFgfb1969Belum ada peringkat
- Research StatementDokumen5 halamanResearch StatementEmad AbdurasulBelum ada peringkat
- Non Linear Root FindingDokumen16 halamanNon Linear Root FindingMichael Ayobami AdelekeBelum ada peringkat
- Applied Mathematics and Computation: Boying Wu, Xiuying LiDokumen4 halamanApplied Mathematics and Computation: Boying Wu, Xiuying LisamiabushnaqBelum ada peringkat
- Sensores ElectroquímicosDokumen5 halamanSensores ElectroquímicosMarcos Reyes VillaBelum ada peringkat
- Theorist's Toolkit Lecture 2-3: LP DualityDokumen9 halamanTheorist's Toolkit Lecture 2-3: LP DualityJeremyKunBelum ada peringkat
- Lab 5: Nonlinear Systems: GoalsDokumen5 halamanLab 5: Nonlinear Systems: GoalsVinodhini RavikumarBelum ada peringkat
- Linear Programming With Two Variables Per Inequality in Poly-Log TimeDokumen16 halamanLinear Programming With Two Variables Per Inequality in Poly-Log TimeDarrelBelum ada peringkat
- Markov Chain and Its Applications (Linear Algebra Applications)Dokumen10 halamanMarkov Chain and Its Applications (Linear Algebra Applications)Xinye YangBelum ada peringkat
- Estimating Definite Integrals: AcknowledgmentDokumen4 halamanEstimating Definite Integrals: AcknowledgmentSai GokulBelum ada peringkat
- AssignmentsDokumen10 halamanAssignmentsDQZBelum ada peringkat
- Numerical Methods: Marisa Villano, Tom Fagan, Dave Fairburn, Chris Savino, David Goldberg, Daniel RaveDokumen44 halamanNumerical Methods: Marisa Villano, Tom Fagan, Dave Fairburn, Chris Savino, David Goldberg, Daniel Raveanirbanpwd76Belum ada peringkat
- M442, Fall 2017 Practice Problems For The MidtermDokumen13 halamanM442, Fall 2017 Practice Problems For The MidtermArun Kumar YadavBelum ada peringkat
- Nithin K Rajendran July-NovemberDokumen4 halamanNithin K Rajendran July-NovembernithinBelum ada peringkat
- Mathematics of Sparsity (And A Few Other Things) : Emmanuel Cand' EsDokumen27 halamanMathematics of Sparsity (And A Few Other Things) : Emmanuel Cand' EsSrinivasaBelum ada peringkat
- 2000 KimDokumen17 halaman2000 KimRaluca-Maria SălcudeanBelum ada peringkat
- Introduction To MultifractalsDokumen23 halamanIntroduction To MultifractalsAnkit Gupta100% (1)
- Wave EquationsDokumen41 halamanWave Equationseren-jaegerBelum ada peringkat
- UntitledDokumen498 halamanUntitledAzarudheen S StatisticsBelum ada peringkat
- Solving The Hamiltonian Cycle Problem Using Symbolic DeterminantsDokumen12 halamanSolving The Hamiltonian Cycle Problem Using Symbolic Determinantstapas_bayen9388Belum ada peringkat
- A Solution To The 3x + 1 ProblemDokumen42 halamanA Solution To The 3x + 1 ProblemPulak RoyBelum ada peringkat
- SF2521NPDE lecture2AKDokumen66 halamanSF2521NPDE lecture2AKBlooD LOVERBelum ada peringkat
- CHE 492 HW 3 Abdulaziz AlhoutiDokumen8 halamanCHE 492 HW 3 Abdulaziz AlhoutiTimelessBelum ada peringkat
- Chap2 603Dokumen29 halamanChap2 603naefmubarak100% (1)
- Diffusion Equation PDFDokumen33 halamanDiffusion Equation PDFDuong Quang DucBelum ada peringkat
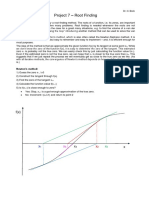
- Project 7 - Root Finding: Newton's MethodDokumen4 halamanProject 7 - Root Finding: Newton's Methodhannan1993Belum ada peringkat
- 6 PDFDokumen5 halaman6 PDFAlawy AbdoBelum ada peringkat
- Full Name of submitter: Cao Thị Minh Anh Student ID: IELSIU18002Dokumen7 halamanFull Name of submitter: Cao Thị Minh Anh Student ID: IELSIU18002ManhBelum ada peringkat
- InequalitiesDokumen38 halamanInequalitiesGabi PetrusBelum ada peringkat
- Lifting Ell - Q-Optimization ThresholdsDokumen29 halamanLifting Ell - Q-Optimization Thresholdscrocoali000Belum ada peringkat
- Reservoir SimulationDokumen4 halamanReservoir Simulationbadjaguar007Belum ada peringkat
- Real Analytic Functions PDFDokumen7 halamanReal Analytic Functions PDFGeorge Mpantes mathematics teacherBelum ada peringkat
- Class 1, 18.05 Jeremy Orloff and Jonathan Bloom 1 Probability vs. StatisticsDokumen20 halamanClass 1, 18.05 Jeremy Orloff and Jonathan Bloom 1 Probability vs. StatisticsNonameBelum ada peringkat
- S. Catterall Et Al - Baby Universes in 4d Dynamical TriangulationDokumen9 halamanS. Catterall Et Al - Baby Universes in 4d Dynamical TriangulationGijke3Belum ada peringkat
- NASA Regression LectureDokumen268 halamanNASA Regression Lecturegustavo rodriguezBelum ada peringkat
- DoyleSnell Random Walks and Electrict NetworksDokumen118 halamanDoyleSnell Random Walks and Electrict NetworksEmmaTeglingBelum ada peringkat
- (E. López-Sandoval) Power Series KineticsDokumen17 halaman(E. López-Sandoval) Power Series KineticsShabbir ChaudharyBelum ada peringkat
- Optimizations For The CSE Machine: Programming Language PrinciplesDokumen12 halamanOptimizations For The CSE Machine: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- Standardizing RPAL AST's: Programming Language PrinciplesDokumen30 halamanStandardizing RPAL AST's: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- The CSE Machine: Programming Language PrinciplesDokumen22 halamanThe CSE Machine: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- Recursion and Fixed-Point Theory: Programming Language PrinciplesDokumen26 halamanRecursion and Fixed-Point Theory: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- Lambda Calculus: Programming Language PrinciplesDokumen26 halamanLambda Calculus: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- The RPAL Functional LanguageDokumen48 halamanThe RPAL Functional LanguageMrunal RuikarBelum ada peringkat
- Writing RPAL Programs: Programming Language PrinciplesDokumen23 halamanWriting RPAL Programs: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- Introduction and Paradigms: Programming Language PrinciplesDokumen19 halamanIntroduction and Paradigms: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- Building AST's For RPAL Programs: Programming Language PrinciplesDokumen15 halamanBuilding AST's For RPAL Programs: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- AST Generation: Programming Language PrinciplesDokumen21 halamanAST Generation: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- Tree Generation: Programming Language PrinciplesDokumen26 halamanTree Generation: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- LL (1) Parsing: Programming Language PrinciplesDokumen21 halamanLL (1) Parsing: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- Parsing: Programming Language PrinciplesDokumen33 halamanParsing: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- Overview of Compilation: Programming Language PrinciplesDokumen28 halamanOverview of Compilation: Programming Language PrinciplesMrunal RuikarBelum ada peringkat
- Gayle McDowell CareerCup Sample ResumeDokumen2 halamanGayle McDowell CareerCup Sample ResumeMrunal RuikarBelum ada peringkat
- Examf99 PsDokumen2 halamanExamf99 PsMrunal RuikarBelum ada peringkat
- Notes On Divide-and-Conquer and Dynamic Programming.: 1 N 1 n/2 n/2 +1 NDokumen11 halamanNotes On Divide-and-Conquer and Dynamic Programming.: 1 N 1 n/2 n/2 +1 NMrunal RuikarBelum ada peringkat
- Operating Systems: Thanks For Downloading. Enjoy The ReadingDokumen9 halamanOperating Systems: Thanks For Downloading. Enjoy The ReadingMrunal RuikarBelum ada peringkat
- Thanks For Downloading. Enjoy The ReadingDokumen15 halamanThanks For Downloading. Enjoy The ReadingMrunal RuikarBelum ada peringkat
- p2p Streaming Short McompDokumen35 halamanp2p Streaming Short McompMrunal RuikarBelum ada peringkat
- Advantages Relative To Other Search AlgorithmsDokumen7 halamanAdvantages Relative To Other Search AlgorithmsMrunal RuikarBelum ada peringkat
- Bloom Filter: Algorithm DescriptionDokumen11 halamanBloom Filter: Algorithm DescriptionMrunal RuikarBelum ada peringkat
- Suf TreeDokumen6 halamanSuf TreeMrunal RuikarBelum ada peringkat
- What Is Javascript?: Thanks For Downloading. Enjoy The ReadingDokumen24 halamanWhat Is Javascript?: Thanks For Downloading. Enjoy The ReadingMrunal RuikarBelum ada peringkat
- Heap TreesDokumen3 halamanHeap TreesNaomi BelonioBelum ada peringkat
- Trachtenberg System - Wikipedia, The Free EncyclopediaDokumen9 halamanTrachtenberg System - Wikipedia, The Free Encyclopediamtf4uBelum ada peringkat
- Siemens Data Sheet 3UF7010-1AU00-0Dokumen10 halamanSiemens Data Sheet 3UF7010-1AU00-0bleerBelum ada peringkat
- Answer All Questions in This SectionDokumen9 halamanAnswer All Questions in This SectionSathis KumarBelum ada peringkat
- Diffie-Hellman Key ExchangeDokumen29 halamanDiffie-Hellman Key ExchangeEswin AngelBelum ada peringkat
- Manual Scilab-5.1.1 PT BRDokumen2.299 halamanManual Scilab-5.1.1 PT BRsilviolhBelum ada peringkat
- Linear Dependence and SpamDokumen5 halamanLinear Dependence and SpamcrisandyBelum ada peringkat
- Quick Help For EDI SEZ IntegrationDokumen2 halamanQuick Help For EDI SEZ IntegrationsrinivasBelum ada peringkat
- Operations of RAEDokumen1 halamanOperations of RAEMargarette SeguerraBelum ada peringkat
- Honeycomb 3d PrintingDokumen12 halamanHoneycomb 3d PrintingNAVEEN SWAMIBelum ada peringkat
- Y2k Project Work On Emr (New)Dokumen114 halamanY2k Project Work On Emr (New)Otunuya Chukwudi Henry EngrBelum ada peringkat
- Sub Net Mask Cheat SheetDokumen3 halamanSub Net Mask Cheat SheetjorgelanfrediBelum ada peringkat
- Rasandik EngineeringDokumen11 halamanRasandik EngineeringelectronicssucksBelum ada peringkat
- Emerging Technology Workshop COLADokumen3 halamanEmerging Technology Workshop COLARaj Daniel Magno58% (12)
- PTA Approved TACS - 28th NovDokumen182 halamanPTA Approved TACS - 28th NovRohit BudhwaniBelum ada peringkat
- 12th Computer Science 1 Mark Questions emDokumen135 halaman12th Computer Science 1 Mark Questions emsathiyaBelum ada peringkat
- Parametric Sweeps in AdsDokumen7 halamanParametric Sweeps in AdssukusportyBelum ada peringkat
- Oracle Database Administration - Checklist Application ReportDokumen27 halamanOracle Database Administration - Checklist Application ReportmathulelrBelum ada peringkat
- Welcome To Excel VBA Eas1Dokumen115 halamanWelcome To Excel VBA Eas1Shailesh KumarBelum ada peringkat
- Alcatel Definitive System Guide PDFDokumen12 halamanAlcatel Definitive System Guide PDFSamuel Mares PalafoxBelum ada peringkat
- Ebook What Is HPCDokumen25 halamanEbook What Is HPCKamenPopovBelum ada peringkat
- Ocfs Oracle Cluster Filesystem For Linux: Users GuideDokumen30 halamanOcfs Oracle Cluster Filesystem For Linux: Users GuideShahid Mahmud100% (1)
- Java Notes For Class IDokumen5 halamanJava Notes For Class ISudipti TiwariBelum ada peringkat
- AP Calculus AB Review Chapter 1Dokumen21 halamanAP Calculus AB Review Chapter 1Preeti RatnamBelum ada peringkat
- Ipx S@e Manager GBDokumen234 halamanIpx S@e Manager GBDuda Misic100% (3)
- Walther Von Der Vogelweide - PoemsDokumen197 halamanWalther Von Der Vogelweide - PoemsYvo YeomansBelum ada peringkat
- BW4HANA Golden Deck 062018 Black JUDokumen74 halamanBW4HANA Golden Deck 062018 Black JUkannanleo67% (3)
- Evaluating One Variable Expressions: Evaluate Each Expression Using The Value GivenDokumen2 halamanEvaluating One Variable Expressions: Evaluate Each Expression Using The Value GivenAmmar RizwanBelum ada peringkat
- Queuing AnalysisDokumen47 halamanQueuing AnalysisAwoke Meskir100% (1)
- Concept Evolution Foundation - 1.2Dokumen277 halamanConcept Evolution Foundation - 1.2abre1get522175% (4)