Anda mungkin juga menyukai
- Monografia LINFOMAS 2Dokumen13 halamanMonografia LINFOMAS 2Claudia TrujilloBelum ada peringkat
- No HodkinDokumen20 halamanNo HodkinDavidBelum ada peringkat
- Sara Baret, FD-1778, Tarea Unidad 3Dokumen13 halamanSara Baret, FD-1778, Tarea Unidad 3Sara Baret PáezBelum ada peringkat
- Todo LeucemiaDokumen42 halamanTodo Leucemiamijaiel_xyzBelum ada peringkat
- Linfomas G2borrarDokumen11 halamanLinfomas G2borrarYemina T ValdesBelum ada peringkat
- LINFOMADokumen13 halamanLINFOMAWillmary MatheusBelum ada peringkat
- Linfoproliferativo CompletoDokumen13 halamanLinfoproliferativo Completoarios_409594Belum ada peringkat
- LINFOMA DE HODGKIN: CARACTERÍSTICAS, CLASIFICACIÓN E IMPLICACIONESDokumen20 halamanLINFOMA DE HODGKIN: CARACTERÍSTICAS, CLASIFICACIÓN E IMPLICACIONESRosales Cruzalegui K. MikaelBelum ada peringkat
- Mieloma MultipleDokumen45 halamanMieloma MultipleLuis RojasBelum ada peringkat
- 66 Neoplasias Malignas LinfoidesDokumen13 halaman66 Neoplasias Malignas LinfoidesKitty Evelyn LooBelum ada peringkat
- Linfoma de HodgkinDokumen5 halamanLinfoma de HodgkinJohan Taborda SierraBelum ada peringkat
- Linfocitoma Cutis. Reporte de Un CasoDokumen5 halamanLinfocitoma Cutis. Reporte de Un CasoMaria PerezBelum ada peringkat
- GUIA2012 LinfomasCelulasManto 2Dokumen17 halamanGUIA2012 LinfomasCelulasManto 2hematologia hgmBelum ada peringkat
- Resumen de Catedra de HEMATO 2Dokumen26 halamanResumen de Catedra de HEMATO 2Catalina MoyaBelum ada peringkat
- Síndromes hematológicosDokumen9 halamanSíndromes hematológicosLisbeth AmparoBelum ada peringkat
- Tercer Parcial Repaso Hema PDFDokumen39 halamanTercer Parcial Repaso Hema PDFabbyBelum ada peringkat
- Patología Del Sistema LinfahematopoyéticoDokumen7 halamanPatología Del Sistema LinfahematopoyéticoJoséDaniel PerdomoBelum ada peringkat
- RESUMEN GENÉTICA - 6.4 y 6.5Dokumen7 halamanRESUMEN GENÉTICA - 6.4 y 6.5Cindy RuizBelum ada peringkat
- Linfooma Del MantoDokumen12 halamanLinfooma Del MantoLeslye VelezBelum ada peringkat
- 8 - Patología de La Medula OseaDokumen12 halaman8 - Patología de La Medula OseaVeronica MendozaBelum ada peringkat
- Leucemia Linfocítica CrónicaDokumen30 halamanLeucemia Linfocítica CrónicaDiego Osvaldo Durán ValadezBelum ada peringkat
- Linfoma No Hodgkin Nunez y OcegueraDokumen34 halamanLinfoma No Hodgkin Nunez y Oceguerajon1224Belum ada peringkat
- CITOLOGIA GANGLIO LINFATICOv2020Dokumen92 halamanCITOLOGIA GANGLIO LINFATICOv2020Army K.UBelum ada peringkat
- Fisiopatologia de Las Leucemias y LinfomasDokumen46 halamanFisiopatologia de Las Leucemias y LinfomasJHUD ARELI PRUDENCIO CHAPABelum ada peringkat
- Neoplasias LinfoidesDokumen62 halamanNeoplasias LinfoidesHever Varon MurciaBelum ada peringkat
- 3 Linfoma de HodgkinDokumen34 halaman3 Linfoma de Hodgkinkevin urillasBelum ada peringkat
- Linfoma No HodkingDokumen6 halamanLinfoma No HodkingAndreaBelum ada peringkat
- Hematopato PDFDokumen37 halamanHematopato PDFManuel HurtadoBelum ada peringkat
- Bolo 13h Linfoma LT (71) 2023Dokumen71 halamanBolo 13h Linfoma LT (71) 2023Rosa Butron HerediaBelum ada peringkat
- Leucemias AgudasDokumen53 halamanLeucemias AgudasJesus GomezBelum ada peringkat
- Patologia Cuaderno 2023-299-305Dokumen7 halamanPatologia Cuaderno 2023-299-305isa suziganBelum ada peringkat
- Linfomas-Camila ArauzDokumen5 halamanLinfomas-Camila ArauzCamila ArauzBelum ada peringkat
- LeucopoyesisDokumen65 halamanLeucopoyesisVanessa DahmerBelum ada peringkat
- Linfomas no Hodgkin: principales subtipos y característicasDokumen6 halamanLinfomas no Hodgkin: principales subtipos y característicasWill MHBelum ada peringkat
- Alteraciones Paraclinicas en LeucemiasDokumen6 halamanAlteraciones Paraclinicas en LeucemiasDiego Ivan EsparzaBelum ada peringkat
- c6 Patologia Del Sistema HematopoyeticoDokumen25 halamanc6 Patologia Del Sistema HematopoyeticochwbaccaBelum ada peringkat
- Enfermedades LinfotericularesDokumen27 halamanEnfermedades LinfotericularesVic motoBelum ada peringkat
- 6.clase de LinfomasDokumen8 halaman6.clase de LinfomasyuniorBelum ada peringkat
- Leucemia Linfática CrónicaDokumen14 halamanLeucemia Linfática Crónicaarianna damaris lopezBelum ada peringkat
- Linfoma no Hodgkin: Características, clasificación y tratamientoDokumen68 halamanLinfoma no Hodgkin: Características, clasificación y tratamientoEdithRuelasRodriguezBelum ada peringkat
- LINFOPROLIFERATIVODokumen30 halamanLINFOPROLIFERATIVODaniel FilhoBelum ada peringkat
- Clase Complementaria Sangre 2021Dokumen10 halamanClase Complementaria Sangre 2021caceres cristianBelum ada peringkat
- Tema 29Dokumen14 halamanTema 29lalalien20Belum ada peringkat
- Leucemias AgudasDokumen87 halamanLeucemias AgudasJuan Lopez100% (1)
- Tratamiento de La Leucemia Mieloide Crónica: Avances FarmacológicosDokumen3 halamanTratamiento de La Leucemia Mieloide Crónica: Avances FarmacológicosJOS LUIS ALANOCA QUISPEBelum ada peringkat
- Caracteristicas BioquimicasDokumen39 halamanCaracteristicas BioquimicasNandor CastilloBelum ada peringkat
- Caso clínico de niña con fiebre, trombocitopenia y monocitosisDokumen5 halamanCaso clínico de niña con fiebre, trombocitopenia y monocitosisJhoel Velásquez BernardoBelum ada peringkat
- Día 1Dokumen44 halamanDía 1Sara Soria EstrugoBelum ada peringkat
- PREGUNTAS SEGUNDO EXAMEN (Neoplasia y Transtornos Hemodinamicos)Dokumen11 halamanPREGUNTAS SEGUNDO EXAMEN (Neoplasia y Transtornos Hemodinamicos)Brenda Marcela Padilla MangonezBelum ada peringkat
- ONCOLOGÍADokumen12 halamanONCOLOGÍAPedro Zúñiga OchoaBelum ada peringkat
- Transcri SeminarioDokumen13 halamanTranscri SeminarioDAVID EDUARDO QUIROZ VARGASBelum ada peringkat
- Fisiopatologia Trabajo Casos Clinicos 2Dokumen9 halamanFisiopatologia Trabajo Casos Clinicos 2Mayra Esther Barreto SotoBelum ada peringkat
- FIsiopato Neoplasias HematológicasDokumen10 halamanFIsiopato Neoplasias HematológicasJuanBelum ada peringkat
- Linfomas T, Angioinmuno, AnaplasicoDokumen53 halamanLinfomas T, Angioinmuno, AnaplasicoMaryu Herrera BustamanteBelum ada peringkat
- Linfoma Folicular 1Dokumen11 halamanLinfoma Folicular 1SHEYLA SHARINA FUENTES PINTOBelum ada peringkat
- Casos Clínico-CitológicosDokumen21 halamanCasos Clínico-CitológicosMiguel Angel Carrasco MedinaBelum ada peringkat
- Genéticas en LinfomasDokumen26 halamanGenéticas en LinfomasJuanCarlosHernandezBalcazarBelum ada peringkat
- Linfomas: definición, clasificación y manifestaciones clínicasDokumen4 halamanLinfomas: definición, clasificación y manifestaciones clínicasSarai VilteBelum ada peringkat
- Neoplasias linfoides: clasificación y característicasDokumen29 halamanNeoplasias linfoides: clasificación y característicasErnesto CabreraBelum ada peringkat
- Absorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleDari EverandAbsorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleBelum ada peringkat
- Demencias Cuerpos de Lewy ExpoDokumen15 halamanDemencias Cuerpos de Lewy ExpomotellawiBelum ada peringkat
- Presentacion de Linfomas BDokumen10 halamanPresentacion de Linfomas BmotellawiBelum ada peringkat
- Transtorno de Movimiento Ii - Miranda Macavilca, GeorgeDokumen14 halamanTranstorno de Movimiento Ii - Miranda Macavilca, GeorgemotellawiBelum ada peringkat
- Análisis de CasoDokumen6 halamanAnálisis de CasomotellawiBelum ada peringkat
- Llc-Miranda Macavilca, GeorgeDokumen8 halamanLlc-Miranda Macavilca, GeorgemotellawiBelum ada peringkat
- VPH: Principales aspectos del virus del papiloma humanoDokumen29 halamanVPH: Principales aspectos del virus del papiloma humanomotellawiBelum ada peringkat
- Factores de Cancer de ColonDokumen9 halamanFactores de Cancer de Colon11-MH-HU-GEORGE CRISTIANI MIRANDA MACAVILCABelum ada peringkat
- Pancreatitis Eosinófila Intersticial: Diagnóstico por ImagenDokumen14 halamanPancreatitis Eosinófila Intersticial: Diagnóstico por ImagenmotellawiBelum ada peringkat
- Puncion Arterial 1Dokumen1 halamanPuncion Arterial 1motellawiBelum ada peringkat
- Artritis ReumatoideDokumen6 halamanArtritis ReumatoidemotellawiBelum ada peringkat
- Epoc ExaDokumen1 halamanEpoc ExamotellawiBelum ada peringkat
- Puncion Arterial PDFDokumen1 halamanPuncion Arterial PDFmotellawiBelum ada peringkat
- Expo FibrosisDokumen9 halamanExpo FibrosismotellawiBelum ada peringkat
- Puncion Arterial 1Dokumen1 halamanPuncion Arterial 1motellawiBelum ada peringkat
- Epoc ExaDokumen1 halamanEpoc ExamotellawiBelum ada peringkat
- Expo Anemia PropeDokumen13 halamanExpo Anemia PropemotellawiBelum ada peringkat
- Interpretación de gasometríaDokumen1 halamanInterpretación de gasometríamotellawiBelum ada peringkat
- Atrapamiento AereoDokumen26 halamanAtrapamiento Aereomotellawi50% (2)
- Estructura básica historia clínica médicaDokumen1 halamanEstructura básica historia clínica médicamotellawiBelum ada peringkat
- Artritis ReumatoideDokumen6 halamanArtritis ReumatoidemotellawiBelum ada peringkat
- Exposicion de Ecologia 2Dokumen16 halamanExposicion de Ecologia 2motellawiBelum ada peringkat
- Fisiopatología de La Anosmia y Disgusia Del Covid - 19Dokumen2 halamanFisiopatología de La Anosmia y Disgusia Del Covid - 19motellawiBelum ada peringkat
- Reglamento-Internado-2019 UDokumen11 halamanReglamento-Internado-2019 UmotellawiBelum ada peringkat
- ANTRODokumen6 halamanANTROmotellawi0% (1)
- Seminario 07 - Famarcalogia EspecialDokumen17 halamanSeminario 07 - Famarcalogia EspecialmotellawiBelum ada peringkat
- Farmaco DobutaminaDokumen20 halamanFarmaco DobutaminamotellawiBelum ada peringkat
- La fatiga mental: un desafío para el bienestar del personal de saludDokumen10 halamanLa fatiga mental: un desafío para el bienestar del personal de saludmotellawiBelum ada peringkat
- Salazar Goicochea & Vargas FustamanteDokumen62 halamanSalazar Goicochea & Vargas FustamantemotellawiBelum ada peringkat
- Historia Clinica, Miranda Macavilca, GeorgeDokumen18 halamanHistoria Clinica, Miranda Macavilca, GeorgemotellawiBelum ada peringkat
- PCR Ultrasensible Turbitest Aa SPDokumen3 halamanPCR Ultrasensible Turbitest Aa SPmotellawi100% (2)
- Repaso Biología T Segundo ParcialDokumen7 halamanRepaso Biología T Segundo ParcialYasmeiky ShawBelum ada peringkat
- Eikenella CorrodensDokumen14 halamanEikenella CorrodensNinoska ValeriaBelum ada peringkat
- CIENCIAS - 8B Santillana Siempre Contigo 2017 PDFDokumen26 halamanCIENCIAS - 8B Santillana Siempre Contigo 2017 PDFJudithGajardoBelum ada peringkat
- Ensayo II de QuimicaDokumen36 halamanEnsayo II de QuimicaIan FloresBelum ada peringkat
- Celula VegetalDokumen27 halamanCelula VegetalMónica CuevasBelum ada peringkat
- Genética en la escuelaDokumen17 halamanGenética en la escuelaXavier Guaillas MorochoBelum ada peringkat
- Guía de Aprendizaje Autónomo Semana 1 La Célula 1.1Dokumen8 halamanGuía de Aprendizaje Autónomo Semana 1 La Célula 1.1Maria Jose Mancilla QuinteroBelum ada peringkat
- UT122Dokumen12 halamanUT122giosue ne0% (1)
- Biologa Cuarto AoDokumen130 halamanBiologa Cuarto AoseemmartBelum ada peringkat
- P424204A1033Dokumen7 halamanP424204A1033jose olayaBelum ada peringkat
- Biologia Practica 4Dokumen10 halamanBiologia Practica 4XIOMARA ANTONELLA CASTREJON RAMOSBelum ada peringkat
- Reino Monera: los procariotasDokumen58 halamanReino Monera: los procariotasHugo ReyesBelum ada peringkat
- Plata ColoidalDokumen6 halamanPlata Coloidalkayvan16Belum ada peringkat
- Informe BioloDokumen4 halamanInforme BioloMonica JimenezBelum ada peringkat
- TRABAJO PRACTICO La Clula y Sus PartesDokumen11 halamanTRABAJO PRACTICO La Clula y Sus PartesYaz JuarezBelum ada peringkat
- Trabajo de Investigacion, Grupo 1 CelulasDokumen12 halamanTrabajo de Investigacion, Grupo 1 CelulasMatias InsfranBelum ada peringkat
- Hepatocitos 1 IDokumen18 halamanHepatocitos 1 ITatianaMachacaBelum ada peringkat
- Lec 21 Anciano SanoDokumen14 halamanLec 21 Anciano SanoGabriela YuseliBelum ada peringkat
- Informe 2 MicrobiologiaDokumen9 halamanInforme 2 MicrobiologiaNatalia PiñonesBelum ada peringkat
- Actividad 7Dokumen3 halamanActividad 7elpanafran2023Belum ada peringkat
- Virus y BacteriasDokumen41 halamanVirus y BacteriasCami Chacon100% (2)
- Cuadro Comparativo de La Clasificación de Los Seres VivosDokumen3 halamanCuadro Comparativo de La Clasificación de Los Seres VivosCarolina Torres83% (24)
- Membrana Celular CeDokumen3 halamanMembrana Celular CeCarla MonteroBelum ada peringkat
- Apuntes de BacteriologiaDokumen36 halamanApuntes de BacteriologiaNiza Martínez CastellanosBelum ada peringkat
- (PDF) 1° SEC. EDA 1 SEMANA 3 CYT 2023 Indagamos Sobre Los Microorganismos Que Habitan en El Agua EstancadaDokumen10 halaman(PDF) 1° SEC. EDA 1 SEMANA 3 CYT 2023 Indagamos Sobre Los Microorganismos Que Habitan en El Agua EstancadaMary Carmen Santa Cruz Alarcon100% (3)
- Recomendaciones para presentar informes de laboratorioDokumen5 halamanRecomendaciones para presentar informes de laboratorioCésar Andrés GómezBelum ada peringkat
- II Parcial PatologiaDokumen2 halamanII Parcial PatologiaBlas Cuba YiyoBelum ada peringkat
- Taller 4 8vo.Dokumen4 halamanTaller 4 8vo.Marisol SiguenzaBelum ada peringkat
- Cuadro Comparativo Clasificacion de Microrganismos DIAMDokumen2 halamanCuadro Comparativo Clasificacion de Microrganismos DIAMDeisy AlvarezBelum ada peringkat
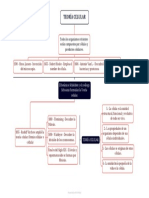
- Tema 1 - Mapa Conceptual - Teoría CelularDokumen1 halamanTema 1 - Mapa Conceptual - Teoría CelularLuisBelum ada peringkat