Anda mungkin juga menyukai
- Other BSM Searches at The LHC: D.Bourilkov (For The ATLAS and CMS Collaborations)Dokumen9 halamanOther BSM Searches at The LHC: D.Bourilkov (For The ATLAS and CMS Collaborations)bourilkovBelum ada peringkat
- Exercise B2 Beam Vibrations: Authors: Submitted (Date) : 2019-09-18 Approved by (Name/date)Dokumen9 halamanExercise B2 Beam Vibrations: Authors: Submitted (Date) : 2019-09-18 Approved by (Name/date)xiaoqi wangBelum ada peringkat
- Final Report Proposal 6-04-252Dokumen3 halamanFinal Report Proposal 6-04-252Yousfi MohamedBelum ada peringkat
- Abstr DRC12Dokumen2 halamanAbstr DRC12smar.marshalBelum ada peringkat
- Technical Note AllpileDokumen1 halamanTechnical Note AllpileDavidBelum ada peringkat
- Nnano 2015 97Dokumen7 halamanNnano 2015 97Pagal KumariBelum ada peringkat
- Modelling Processes in - SANS AnalysisDokumen1 halamanModelling Processes in - SANS Analysiskorkeakoulu5984Belum ada peringkat
- Poster - 副本Dokumen1 halamanPoster - 副本Ke LiBelum ada peringkat
- STU - S6 Opt - GA - : Université Cadi Ayyad Faculté Des Sciences SemlaliaDokumen3 halamanSTU - S6 Opt - GA - : Université Cadi Ayyad Faculté Des Sciences SemlaliaBrahim AIT BENALIBelum ada peringkat
- Công Ty Cổ Phần Đầu Tư Bất Động SảnDokumen9 halamanCông Ty Cổ Phần Đầu Tư Bất Động SảnHÙNG VĨ LANDBelum ada peringkat
- Advances in Quantitative XRD Analysis For ClinkerDokumen11 halamanAdvances in Quantitative XRD Analysis For Clinkervũ minh tâmBelum ada peringkat
- Mobility-Stability Trade-Off in Oxide Thin-Film Transistors: ArticlesDokumen8 halamanMobility-Stability Trade-Off in Oxide Thin-Film Transistors: Articles禾六安Belum ada peringkat
- (Kurt J. Lester, 2018) Pressure Measurement Technical NotesDokumen5 halaman(Kurt J. Lester, 2018) Pressure Measurement Technical NotesLucelly Muñoz AmarilesBelum ada peringkat
- Cellular Concrete With Addition of Aluminum Recycled Foil PowdersDokumen11 halamanCellular Concrete With Addition of Aluminum Recycled Foil PowdersRaah E SulookBelum ada peringkat
- ATMS 564 Lecture wk1Dokumen14 halamanATMS 564 Lecture wk1Kakashi 12Belum ada peringkat
- 03 Wave TheoryDokumen19 halaman03 Wave TheoryAashishBelum ada peringkat
- Front Mulcher Specifications and Features for Skid Steer and Wheel Loaders Under 40 CharactersDokumen1 halamanFront Mulcher Specifications and Features for Skid Steer and Wheel Loaders Under 40 CharactersKft. Progép-TechnikaBelum ada peringkat
- Advances in Quantitative XRD Analysis For ClinkerDokumen11 halamanAdvances in Quantitative XRD Analysis For ClinkerAdriano CarvalhoBelum ada peringkat
- CellTiter 96 AQueous One Solution Cell Proliferation Assay TB245Dokumen13 halamanCellTiter 96 AQueous One Solution Cell Proliferation Assay TB245Teh Chye PhingBelum ada peringkat
- SPE 154567 Polymer Flooding A 500-cp OilDokumen13 halamanSPE 154567 Polymer Flooding A 500-cp OilBichara DjimetBelum ada peringkat
- Rapid mining development through tunneling techniquesDokumen3 halamanRapid mining development through tunneling techniquesDamian SaldarriagaBelum ada peringkat
- Evaluation & Rehabilitation of An Abandoned Mining SiteDokumen24 halamanEvaluation & Rehabilitation of An Abandoned Mining SitetonmoyBelum ada peringkat
- Rock Physchics of PorosityDokumen6 halamanRock Physchics of PorosityMirza SadewaBelum ada peringkat
- 1 - Gulfstream Park PPDokumen31 halaman1 - Gulfstream Park PPcanaimaapolo2023Belum ada peringkat
- Nphoton 2011 15Dokumen8 halamanNphoton 2011 15王子昱Belum ada peringkat
- M.techDokumen15 halamanM.techsonu kumarBelum ada peringkat
- Research Statement: Nicholas J. MooreDokumen5 halamanResearch Statement: Nicholas J. MooreValdir JúniorBelum ada peringkat
- Poster 1Dokumen1 halamanPoster 1docteur besghaierBelum ada peringkat
- Introduction to Semiconductors: Understanding Valence and Conduction BandsDokumen16 halamanIntroduction to Semiconductors: Understanding Valence and Conduction BandsBariya Hitesh VijayBelum ada peringkat
- Fluorescence Microscopy Alternative for Evaluating Organic-Inorganic Composite DispersionDokumen7 halamanFluorescence Microscopy Alternative for Evaluating Organic-Inorganic Composite DispersionFlorent MortierBelum ada peringkat
- BF02269611Dokumen9 halamanBF02269611otipicni6969Belum ada peringkat
- On Globally Nilpotent Differential Equations: Michael Dettweiler and Stefan Reiter July 20, 2006Dokumen15 halamanOn Globally Nilpotent Differential Equations: Michael Dettweiler and Stefan Reiter July 20, 2006skrybantBelum ada peringkat
- Graphene Plasmonics For Tunable Terahertz MetamaterialsDokumen5 halamanGraphene Plasmonics For Tunable Terahertz MetamaterialsHabibe DurmazBelum ada peringkat
- John Kullman How To Choose The ProppantDokumen55 halamanJohn Kullman How To Choose The ProppantUmmu MuadzBelum ada peringkat
- DI FittingsDokumen1 halamanDI FittingsJhansiBelum ada peringkat
- 4RA34RB3 Lecture Note-8 HPGe DetectorDokumen20 halaman4RA34RB3 Lecture Note-8 HPGe DetectorSlide ShareBelum ada peringkat
- Plant Nutrition QpansDokumen18 halamanPlant Nutrition QpansSuhaan HussainBelum ada peringkat
- Photosynthesis Rates and Light WavelengthDokumen11 halamanPhotosynthesis Rates and Light WavelengthAbdul RehmanBelum ada peringkat
- 2.1 LedDokumen5 halaman2.1 LedFernando Reza CamposBelum ada peringkat
- Im 2014Dokumen6 halamanIm 2014Jhon BurbanoBelum ada peringkat
- 5 Economy Picking Licks: WWW - Jenslarsen.nl Jens LarsenDokumen2 halaman5 Economy Picking Licks: WWW - Jenslarsen.nl Jens LarsenYngwie MalmsteenBelum ada peringkat
- Turning A Molecule Into A Coherent Two-Level Quantum System: ArticlesDokumen9 halamanTurning A Molecule Into A Coherent Two-Level Quantum System: Articlespolar necksonBelum ada peringkat
- SPE 154567 Polymer Flooding A 500-cp OilDokumen13 halamanSPE 154567 Polymer Flooding A 500-cp OilfuatBelum ada peringkat
- Optical Study of PolySiDokumen7 halamanOptical Study of PolySiJohnny CrossBelum ada peringkat
- SPIE ConferenceDokumen6 halamanSPIE ConferenceSeenu IIscBelum ada peringkat
- Modelling of Chloride PenetrationDokumen26 halamanModelling of Chloride PenetrationDamir ZenunovicBelum ada peringkat
- Next Generation Optical Fibers: Challenges and OpportunitiesDokumen4 halamanNext Generation Optical Fibers: Challenges and OpportunitiesMarco AntonioBelum ada peringkat
- Simulation of Short Circuit and Lightning Transients On 110 KV Overhead and Cable Transmission Lines Using ATP-EMTPDokumen14 halamanSimulation of Short Circuit and Lightning Transients On 110 KV Overhead and Cable Transmission Lines Using ATP-EMTPDejanBelum ada peringkat
- Uf1007a02 1Dokumen1 halamanUf1007a02 1MARTIN GONZALEZBelum ada peringkat
- Electrospinning Nanofibers of PANI/PMMA BlendsDokumen6 halamanElectrospinning Nanofibers of PANI/PMMA BlendsDanay ManzoBelum ada peringkat
- Sensing Device For Camless Engine Electromagnetic Actuators: Abstract - A Position Reconstruction Method For Camless enDokumen6 halamanSensing Device For Camless Engine Electromagnetic Actuators: Abstract - A Position Reconstruction Method For Camless enSantosh MsdsBelum ada peringkat
- hanabata1989Dokumen12 halamanhanabata1989xrovljolscjvmiszchBelum ada peringkat
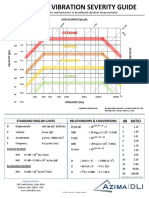
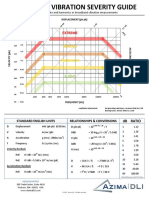
- AzimaDLI Severity Chart 2013 PDFDokumen1 halamanAzimaDLI Severity Chart 2013 PDFEswin Paico de la Cruz100% (1)
- Plastic Shrinkage Cracking (BPS)Dokumen36 halamanPlastic Shrinkage Cracking (BPS)shingkeongBelum ada peringkat
- AzimaDLI Severity Chart 2013 PDFDokumen1 halamanAzimaDLI Severity Chart 2013 PDFFernando BaconBelum ada peringkat
- AzimaDLI Severity Chart 2013 PDFDokumen1 halamanAzimaDLI Severity Chart 2013 PDFPraveen SreedharanBelum ada peringkat
- Ind 0930 PDokumen25 halamanInd 0930 PMichel MichelBelum ada peringkat
- AzimaDLI Severity Chart 2013 PDFDokumen1 halamanAzimaDLI Severity Chart 2013 PDFEswin Paico de la CruzBelum ada peringkat
- Final Report Proposal 6-04-237Dokumen4 halamanFinal Report Proposal 6-04-237Yousfi MohamedBelum ada peringkat
- Final Report Proposal 6-04-237Dokumen4 halamanFinal Report Proposal 6-04-237Yousfi MohamedBelum ada peringkat
- Final Report Proposal 6-04-237Dokumen4 halamanFinal Report Proposal 6-04-237Yousfi MohamedBelum ada peringkat
- Mohamed YousfiDokumen5 halamanMohamed YousfiYousfi MohamedBelum ada peringkat
- Physics Semiconductor Device MCQDokumen3 halamanPhysics Semiconductor Device MCQAsim Ali0% (1)
- Air Cooled Screw Chiller Performance SpecificationDokumen2 halamanAir Cooled Screw Chiller Performance SpecificationDajuko Butarbutar100% (1)
- Sist-En-6101-2016 .Dokumen9 halamanSist-En-6101-2016 .lokelooksBelum ada peringkat
- Mental AspectDokumen29 halamanMental AspectBenjii CarlosBelum ada peringkat
- EN6VC IIIa 6.2 - 2023 2024Dokumen2 halamanEN6VC IIIa 6.2 - 2023 2024Ma. Feliza SaliganBelum ada peringkat
- Ayurveda Signs of LifeDokumen15 halamanAyurveda Signs of LifeSanjeethBelum ada peringkat
- Bicycle Repair ManualDokumen162 halamanBicycle Repair Manualrazvancc89% (9)
- Project Planning HandbookDokumen21 halamanProject Planning HandbookPhilip JonesBelum ada peringkat
- Delivered Voided Application (Surrender Instrument) Returned To at - Sik - Hata Nation of Yamasee MoorsDokumen20 halamanDelivered Voided Application (Surrender Instrument) Returned To at - Sik - Hata Nation of Yamasee MoorsMARK MENO©™Belum ada peringkat
- Curriculum Vitae: Name: Mobile: EmailDokumen3 halamanCurriculum Vitae: Name: Mobile: EmailRākesh RakhiBelum ada peringkat
- PC 4 Product List 2019 - Pc4Dokumen28 halamanPC 4 Product List 2019 - Pc4ShBelum ada peringkat
- The Advantages and Disadvantages If Block ChainDokumen7 halamanThe Advantages and Disadvantages If Block ChainKarthik ShettyBelum ada peringkat
- PLTW: Digital Electronics Syllabus For Mrs. Yusufi: Unit 1: Fundamentals of Analog and DigitalDokumen5 halamanPLTW: Digital Electronics Syllabus For Mrs. Yusufi: Unit 1: Fundamentals of Analog and DigitalTriston DurbinBelum ada peringkat
- Smarter Washing Solutions: Modular Wash RangeDokumen5 halamanSmarter Washing Solutions: Modular Wash RangeSujesh AnBelum ada peringkat
- Mitigating arc ash hazards design constraintsDokumen6 halamanMitigating arc ash hazards design constraintswaqas_a_shaikh4348Belum ada peringkat
- Eccsa Five Year (2014 15 - 2018 19) Strategic PlanDokumen95 halamanEccsa Five Year (2014 15 - 2018 19) Strategic Planyayehyirad100% (1)
- Analisis Efektivitas Inklusi Keuangan Di BMT Syariah Riyal: PendahuluanDokumen25 halamanAnalisis Efektivitas Inklusi Keuangan Di BMT Syariah Riyal: PendahuluanEma Rina SafitriBelum ada peringkat
- Assembly Transmission Volvo A40GDokumen52 halamanAssembly Transmission Volvo A40GNanang SetiawanBelum ada peringkat
- Cropprotectionequipment-Rocker Sprayer - Specification (: Indian StandardDokumen9 halamanCropprotectionequipment-Rocker Sprayer - Specification (: Indian Standardgini associatesBelum ada peringkat
- BS (English) Dept of English University of SargodhaDokumen36 halamanBS (English) Dept of English University of SargodhaFEROZ KHANBelum ada peringkat
- TLUD Handbook, Paul Anderson, V.2010Dokumen19 halamanTLUD Handbook, Paul Anderson, V.2010satyakaamsBelum ada peringkat
- CA Ashish Dewani - Resume-1Dokumen2 halamanCA Ashish Dewani - Resume-1Payal JainBelum ada peringkat
- NCP 1014Dokumen24 halamanNCP 1014rodricaldasBelum ada peringkat
- Overview On Image Captioning TechniquesDokumen6 halamanOverview On Image Captioning TechniquesWARSE JournalsBelum ada peringkat
- Kevin Chiu - Solving Procrastination v1.1Dokumen2 halamanKevin Chiu - Solving Procrastination v1.1TêteHauteBelum ada peringkat
- Carbon SteelDokumen1 halamanCarbon SteeldexterBelum ada peringkat
- Sensor Guide: Standard Triaxial Geophones Specialty Triaxial Geophones Standard Overpressure MicrophonesDokumen1 halamanSensor Guide: Standard Triaxial Geophones Specialty Triaxial Geophones Standard Overpressure MicrophonesDennis Elias TaipeBelum ada peringkat
- DLP IN ICT 9 1st MeetingDokumen2 halamanDLP IN ICT 9 1st MeetingHEDDA FULOBelum ada peringkat
- An Introduction To Pascal Programming MOD 2010Dokumen5 halamanAn Introduction To Pascal Programming MOD 2010Johnas DalusongBelum ada peringkat
- Jupiter - The Giant Planet That Destroys CometsDokumen2 halamanJupiter - The Giant Planet That Destroys Cometsmaiche amarBelum ada peringkat