Anda mungkin juga menyukai
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- MSYS-1 0 11-ChangesDokumen3 halamanMSYS-1 0 11-ChangesCyril BerthelotBelum ada peringkat
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (587)
- The Professional DesktopDokumen318 halamanThe Professional Desktopalintuta2Belum ada peringkat
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (890)
- 0001981572-JAR Resources in JNLP File Are Not Signed by Same CertificateDokumen13 halaman0001981572-JAR Resources in JNLP File Are Not Signed by Same CertificateAnonymous AZGp1KBelum ada peringkat
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- Seksioni I Kabllos Per Rrymat e Lidhjes Se ShkurteDokumen1 halamanSeksioni I Kabllos Per Rrymat e Lidhjes Se ShkurteDukagjin Ramqaj100% (1)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (399)
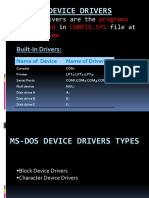
- Ms-Dos Device Drivers: Device Drivers Are The That in File atDokumen13 halamanMs-Dos Device Drivers: Device Drivers Are The That in File atJass GillBelum ada peringkat
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- Ikan Di Kepualauan Indo-AustraliaDokumen480 halamanIkan Di Kepualauan Indo-AustraliaDediBelum ada peringkat
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- CBSE Class 5 Mathematics Sample Paper Set NDokumen4 halamanCBSE Class 5 Mathematics Sample Paper Set NRamanjeet KaurBelum ada peringkat
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Apex Ch10c1 Chassis At2408s Ch04t1002 Om8839ps Tda4605 TV SMDokumen61 halamanApex Ch10c1 Chassis At2408s Ch04t1002 Om8839ps Tda4605 TV SMAlejo Alex CondeBelum ada peringkat
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- EET422 EMC Intro-Banana Skins 2011-2012 MSWDokumen6 halamanEET422 EMC Intro-Banana Skins 2011-2012 MSWVeeradasan PerumalBelum ada peringkat
- Synology DS718 Plus Data Sheet EnuDokumen6 halamanSynology DS718 Plus Data Sheet EnuSteve AttwoodBelum ada peringkat
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- PTP - Level MethodsDokumen23 halamanPTP - Level Methodssasikiran mBelum ada peringkat
- Numeri OrdinaliDokumen2 halamanNumeri OrdinaliClaudioBelum ada peringkat
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- Zarlino-On The ModesDokumen150 halamanZarlino-On The ModesPartituraDireccion100% (1)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- CI SetDokumen18 halamanCI Setতন্ময় ঢালি Tanmay DhaliBelum ada peringkat
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2219)
- Solvent based printing inks applicationsDokumen34 halamanSolvent based printing inks applicationsAmna liaquatBelum ada peringkat
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- VSD Operacion ControlDokumen138 halamanVSD Operacion ControlLeon PerezBelum ada peringkat
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- HowTo Work With CR 90Dokumen87 halamanHowTo Work With CR 90WagBezerraBelum ada peringkat
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (265)
- 3RP15 05-1aDokumen3 halaman3RP15 05-1atycristinaBelum ada peringkat
- Example 3 - S-Beam CrashDokumen13 halamanExample 3 - S-Beam CrashSanthosh LingappaBelum ada peringkat
- WR424GB00DDokumen16 halamanWR424GB00DIgor San Martín PeñalozaBelum ada peringkat
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- Ain 2016 Pilot Report m600Dokumen4 halamanAin 2016 Pilot Report m600Antonio Cesar de Sa LeitaoBelum ada peringkat
- Database Classification TypesDokumen10 halamanDatabase Classification TypesBhiea Mische MatilacBelum ada peringkat
- Bottazzini RiemannDokumen36 halamanBottazzini RiemanncedillaBelum ada peringkat
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- M6 2020 Binomial Distribution Lecture NotesDokumen27 halamanM6 2020 Binomial Distribution Lecture Notescoyite8695Belum ada peringkat
- SERVICE MANUAL CRAWLER EXCAVATOR R210LC-7Dokumen1 halamanSERVICE MANUAL CRAWLER EXCAVATOR R210LC-7DmitryBelum ada peringkat
- Login Form: User Name Password Remember MeDokumen8 halamanLogin Form: User Name Password Remember MeBridget Anne BenitezBelum ada peringkat
- 11+ Entrance Examination: Specimen PaperDokumen8 halaman11+ Entrance Examination: Specimen PaperNayem Hossain HemuBelum ada peringkat
- Wsat200 RamsaDokumen12 halamanWsat200 RamsaAndy ColeBelum ada peringkat
- Max Born, Albert Einstein-The Born-Einstein Letters-Macmillan (1971)Dokumen132 halamanMax Born, Albert Einstein-The Born-Einstein Letters-Macmillan (1971)Brian O'SullivanBelum ada peringkat
- AND Optimization OF Three Existing Ethylbenzene Dehydrogenation Reactors in SeriesDokumen5 halamanAND Optimization OF Three Existing Ethylbenzene Dehydrogenation Reactors in SeriesMuhammad Ridwan TanjungBelum ada peringkat
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (119)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)