Anda mungkin juga menyukai
- Review 402Dokumen50 halamanReview 402Serdar CharyyevBelum ada peringkat
- Conjugated Dyes Lab EditedDokumen8 halamanConjugated Dyes Lab EditedGugu Rutherford100% (1)
- The Diels-Alder ReactionDokumen351 halamanThe Diels-Alder ReactionRay Frausto100% (1)
- Solid State Chapter 2Dokumen75 halamanSolid State Chapter 2Ambreen KhanBelum ada peringkat
- Medicinal Chemistry—III: Main Lectures Presented at the Third International Symposium on Medicinal ChemistryDari EverandMedicinal Chemistry—III: Main Lectures Presented at the Third International Symposium on Medicinal ChemistryP. PratesiBelum ada peringkat
- MSC Chemistry Paper-IX Unit-5Dokumen56 halamanMSC Chemistry Paper-IX Unit-5Alexa Torres100% (1)
- CH2203 - Spectroscopy of Inorganic CompoundsDokumen6 halamanCH2203 - Spectroscopy of Inorganic CompoundsJohnBelum ada peringkat
- Berg Uncertainty PrincipleDokumen20 halamanBerg Uncertainty PrincipleSiddhi Nitin Mahajan0% (1)
- Chapter 4 Analytical MethodsDokumen104 halamanChapter 4 Analytical MethodsCL SanchezBelum ada peringkat
- Lab #1: Absorption Spectra of Conjugated Dyes: E E E EDokumen5 halamanLab #1: Absorption Spectra of Conjugated Dyes: E E E EIreneVeladoBelum ada peringkat
- Physics Experiment ReportDokumen12 halamanPhysics Experiment Reportdreanor100% (5)
- Radioisotope TechniquesDokumen63 halamanRadioisotope TechniquesPallavi Kanungo UpritBelum ada peringkat
- (UV Vis) SpectrosDokumen4 halaman(UV Vis) SpectrosGarion Charles0% (1)
- NMR CouplingDokumen72 halamanNMR CouplingShrutiBelum ada peringkat
- Analytical Chemistry Quiz MaterialDokumen11 halamanAnalytical Chemistry Quiz MaterialAltaf Ur RehmanBelum ada peringkat
- Electrophoreti C Methods: Igaa SeptiariDokumen22 halamanElectrophoreti C Methods: Igaa SeptiariGung Ari100% (1)
- Electric Charge and FieldsDokumen25 halamanElectric Charge and FieldsShrezBelum ada peringkat
- B.SC Organic Chemistry (Paper-5) - Q and ADokumen59 halamanB.SC Organic Chemistry (Paper-5) - Q and ASyed furkhanBelum ada peringkat
- Practical Analytical 1 ,,chemistryDokumen45 halamanPractical Analytical 1 ,,chemistryFadlin AdimBelum ada peringkat
- High Spin and Low Spin ComplexesDokumen13 halamanHigh Spin and Low Spin ComplexesMa'arif A. SyafiiBelum ada peringkat
- Various Tools and Techniques Used For Structural ElucidationDokumen28 halamanVarious Tools and Techniques Used For Structural ElucidationAvinashBelum ada peringkat
- Fourier Transform Infrared SpectrosDokumen27 halamanFourier Transform Infrared Spectrosrmarin_90Belum ada peringkat
- CHEM 430 NMR Spectroscopy Chapter 3Dokumen97 halamanCHEM 430 NMR Spectroscopy Chapter 3Praveen kumar100% (1)
- Applied Chemistry (UCB008) : - Instructor - Dr. Soumen Basu Associate Professor, School of Chemistry and BiochemistryDokumen33 halamanApplied Chemistry (UCB008) : - Instructor - Dr. Soumen Basu Associate Professor, School of Chemistry and BiochemistrySukh SindhiBelum ada peringkat
- Chapter 16 Magnetic Fields: Electricity and MagnetismDokumen11 halamanChapter 16 Magnetic Fields: Electricity and MagnetismeltytanBelum ada peringkat
- Progressive Wave22Dokumen29 halamanProgressive Wave22lianghoo94Belum ada peringkat
- Partial VolumeDokumen38 halamanPartial VolumeJames KuBelum ada peringkat
- Minamata Disease: Mercury PoisoningDokumen18 halamanMinamata Disease: Mercury PoisoningBijay Kumar MahatoBelum ada peringkat
- L2 Che101Dokumen16 halamanL2 Che101Musa Ahammed MahinBelum ada peringkat
- Units and DimensionsDokumen61 halamanUnits and DimensionsPrudhvi RajBelum ada peringkat
- Uv Visible Spectroscopy: by Nandesh V. PingaleDokumen38 halamanUv Visible Spectroscopy: by Nandesh V. PingaleMohammed Adil ShareefBelum ada peringkat
- Molecular Luminescence SpectrosDokumen19 halamanMolecular Luminescence Spectrosedwedq100% (1)
- Rutherford and Bohr's Atomic ModelDokumen101 halamanRutherford and Bohr's Atomic Modelthrivikram100% (1)
- Spectroscopy and ChromatographyDokumen7 halamanSpectroscopy and ChromatographyPa GesBelum ada peringkat
- Iub Pha404 Autumn 2022 Ms BasicDokumen52 halamanIub Pha404 Autumn 2022 Ms BasicTanvir FahimBelum ada peringkat
- Department of Physics Laboratory Manual For Nuclear Physics (Phys2052)Dokumen132 halamanDepartment of Physics Laboratory Manual For Nuclear Physics (Phys2052)Tadesse AbateBelum ada peringkat
- Chapter 19 NMRDokumen126 halamanChapter 19 NMRMuchammad RofiiBelum ada peringkat
- UV Spectroscopy 2016Dokumen87 halamanUV Spectroscopy 2016M Mudassar AslamBelum ada peringkat
- Molecular Spectroscopy: BackgroundDokumen45 halamanMolecular Spectroscopy: Backgroundsavvy_as_98100% (1)
- Basics in NMRDokumen75 halamanBasics in NMRPrashant PandeyBelum ada peringkat
- Infrared Spectrometry: Cua-Narciso-Pilar, Expt 7Dokumen47 halamanInfrared Spectrometry: Cua-Narciso-Pilar, Expt 7Karina NarcisoBelum ada peringkat
- CHM 101 Complete - LNDokumen80 halamanCHM 101 Complete - LNSimon Adediran100% (1)
- Organic Transistors OFTFDokumen30 halamanOrganic Transistors OFTFIsrati ValeriuBelum ada peringkat
- Physics AssignmentDokumen22 halamanPhysics AssignmentVeerareddy Vippala100% (1)
- Biosensors RPCDokumen72 halamanBiosensors RPCSajjad Hossain ShuvoBelum ada peringkat
- Optical SpectrosDokumen7 halamanOptical SpectrosgayaxniBelum ada peringkat
- Atomic Term SymbolsDokumen13 halamanAtomic Term SymbolsAnish RaoBelum ada peringkat
- Nuclear Magnetic Resonance: Half-Integer Odd Odd or EvenDokumen19 halamanNuclear Magnetic Resonance: Half-Integer Odd Odd or EvenRAJ VYASBelum ada peringkat
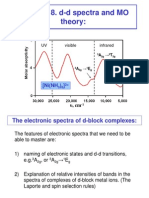
- Chemistry 445 Lecture 18 MO Theory and D-D SpectraDokumen22 halamanChemistry 445 Lecture 18 MO Theory and D-D SpectraAbhinav JainBelum ada peringkat
- Stereochemistry New L1-L3Dokumen107 halamanStereochemistry New L1-L3Pareen5100% (2)
- UV-Vis Spectroscopy: Chm622-Advance Organic SpectrosDokumen50 halamanUV-Vis Spectroscopy: Chm622-Advance Organic Spectrossharifah sakinah syed soffianBelum ada peringkat
- Lab. Conjugated DyesDokumen5 halamanLab. Conjugated DyesIreneVeladoBelum ada peringkat
- Perspectives: A Comparison of RAFT and ATRP Methods For Controlled Radical PolymerizationDokumen11 halamanPerspectives: A Comparison of RAFT and ATRP Methods For Controlled Radical PolymerizationEl MisterioBelum ada peringkat
- Some Basic Concepts in ChemistryDokumen19 halamanSome Basic Concepts in Chemistryg_ayyanarBelum ada peringkat
- 2D NMRDokumen10 halaman2D NMRHariprasad Reddy100% (1)
- Mass Spectrometry: Ev MVDokumen12 halamanMass Spectrometry: Ev MVMuhammad Tariq RazaBelum ada peringkat
- Employee EngagementDokumen3 halamanEmployee EngagementUma Villashini GunasekaranBelum ada peringkat
- Essay - 111Dokumen5 halamanEssay - 111Uma Villashini GunasekaranBelum ada peringkat
- Reference ElectrodesDokumen3 halamanReference ElectrodesprydeepBelum ada peringkat
- CHM 3402 Experiment 8Dokumen11 halamanCHM 3402 Experiment 8Uma Villashini GunasekaranBelum ada peringkat
- CHM 3402 Experiment 6Dokumen7 halamanCHM 3402 Experiment 6Uma Villashini GunasekaranBelum ada peringkat
- Infra Red Spectroscopy (Plus Worksheet Answers)Dokumen47 halamanInfra Red Spectroscopy (Plus Worksheet Answers)lianchen251110Belum ada peringkat
- German Problems 2014Dokumen152 halamanGerman Problems 2014Yosi W KusumaBelum ada peringkat
- SyllabusDokumen2 halamanSyllabusCharles HoBelum ada peringkat
- PPTDokumen46 halamanPPTJafrinta Irma Ruta Astari100% (1)
- Paul Loo - Convenient Synthesis and Spectroscopic Data of Methcathinone AnalogsDokumen27 halamanPaul Loo - Convenient Synthesis and Spectroscopic Data of Methcathinone AnalogsFedrm100% (2)
- Chem 442 - Chapter OneDokumen26 halamanChem 442 - Chapter OneArindam DasBelum ada peringkat
- Structural and Optical Properties of 6,13-Pentacenequinone Thin FilmsDokumen4 halamanStructural and Optical Properties of 6,13-Pentacenequinone Thin FilmsClaudio BiaginiBelum ada peringkat
- Cigarrete Smoke Rotational SpectrosDokumen3 halamanCigarrete Smoke Rotational SpectrosJacqueline FSBelum ada peringkat
- An Overview of Forensic Drug Testing Methods and TDokumen14 halamanAn Overview of Forensic Drug Testing Methods and TTuấn Minh PhùngBelum ada peringkat
- Chemistry 2022 AKTUDokumen2 halamanChemistry 2022 AKTUVicky SrivastavaBelum ada peringkat
- Indira Gandhi Centre Atomic ResearchDokumen8 halamanIndira Gandhi Centre Atomic ResearchTrcStaffBelum ada peringkat
- 4 - SpectrophotometerDokumen54 halaman4 - SpectrophotometerSamad WokyohBelum ada peringkat
- Berlin Photonics and Optics Companies - 5.0Dokumen8 halamanBerlin Photonics and Optics Companies - 5.0Suman RachaBelum ada peringkat
- Atomic Emission SpectrosDokumen16 halamanAtomic Emission SpectrosHamza Ali MinhasBelum ada peringkat
- 19F, 31P, 14N NMRDokumen5 halaman19F, 31P, 14N NMRManoj kumar SahooBelum ada peringkat
- Materials Chemistry and Physics: Herv e K. Tchakoute, Claus H. Rüscher, E. Kamseu, Jean N.Y. Djobo, C. LeonelliDokumen9 halamanMaterials Chemistry and Physics: Herv e K. Tchakoute, Claus H. Rüscher, E. Kamseu, Jean N.Y. Djobo, C. LeonelliAntonio Ernandes Macedo PaivaBelum ada peringkat
- NMR Spectroscopy: by Darshan R. Telange, KNCP, Butibori (Nagpur)Dokumen60 halamanNMR Spectroscopy: by Darshan R. Telange, KNCP, Butibori (Nagpur)team engineer100% (1)
- 10.1007@978 3 030 21638 2Dokumen214 halaman10.1007@978 3 030 21638 2dortiBelum ada peringkat
- Wu 2007Dokumen30 halamanWu 2007gjfhkjBelum ada peringkat
- Applied Physics - 20.7Dokumen35 halamanApplied Physics - 20.7AJESH MBelum ada peringkat
- 1999 - The Elusive Mechanism of The Magnetic Memory of WaterDokumen8 halaman1999 - The Elusive Mechanism of The Magnetic Memory of WaterPat BigorniaBelum ada peringkat
- 4.74 Chemistry PET SyllabusDokumen14 halaman4.74 Chemistry PET SyllabusShifa ChaudhariBelum ada peringkat
- Spektra AtomDokumen21 halamanSpektra AtomIrvandar NurviandyBelum ada peringkat
- Turbidimetry and Nephelometry SDDokumen9 halamanTurbidimetry and Nephelometry SDCamilo Varela VegaBelum ada peringkat
- Surface-Enhanced Raman Scattering in Surface Science: Loreta Garmutė MGTF-7Dokumen19 halamanSurface-Enhanced Raman Scattering in Surface Science: Loreta Garmutė MGTF-7AAAAABelum ada peringkat
- Exp 4 Postlab Worksheet Identification of An Unknown Through Qualitative AnalysisDokumen4 halamanExp 4 Postlab Worksheet Identification of An Unknown Through Qualitative AnalysisKarl CarandangBelum ada peringkat
- OrcaManual 2.7.0Dokumen418 halamanOrcaManual 2.7.0Kristhian Alcantar MedinaBelum ada peringkat
- VX Spectra - JoshuaDokumen1 halamanVX Spectra - JoshuaJoshua SitorusBelum ada peringkat
- Young EinsteinDokumen9 halamanYoung EinsteinSomu ShivaBelum ada peringkat
- NSCI 115: Chemical Principles of NANO I Lab 1: UV-Vis Spectroscopy and Beer-Lambert LawDokumen6 halamanNSCI 115: Chemical Principles of NANO I Lab 1: UV-Vis Spectroscopy and Beer-Lambert LawIsaac SnitkoffBelum ada peringkat
- Giza: The Tesla Connection: Acoustical Science and the Harvesting of Clean EnergyDari EverandGiza: The Tesla Connection: Acoustical Science and the Harvesting of Clean EnergyBelum ada peringkat
- A Brief History of Time: From the Big Bang to Black HolesDari EverandA Brief History of Time: From the Big Bang to Black HolesPenilaian: 4 dari 5 bintang4/5 (2193)
- Dark Matter and the Dinosaurs: The Astounding Interconnectedness of the UniverseDari EverandDark Matter and the Dinosaurs: The Astounding Interconnectedness of the UniversePenilaian: 3.5 dari 5 bintang3.5/5 (69)
- Knocking on Heaven's Door: How Physics and Scientific Thinking Illuminate the Universe and the Modern WorldDari EverandKnocking on Heaven's Door: How Physics and Scientific Thinking Illuminate the Universe and the Modern WorldPenilaian: 3.5 dari 5 bintang3.5/5 (64)
- Higgs Discovery: The Power of Empty SpaceDari EverandHiggs Discovery: The Power of Empty SpacePenilaian: 3 dari 5 bintang3/5 (30)
- Summary and Interpretation of Reality TransurfingDari EverandSummary and Interpretation of Reality TransurfingPenilaian: 5 dari 5 bintang5/5 (5)
- The Magick of Physics: Uncovering the Fantastical Phenomena in Everyday LifeDari EverandThe Magick of Physics: Uncovering the Fantastical Phenomena in Everyday LifeBelum ada peringkat
- A Beginner's Guide to Constructing the Universe: The Mathematical Archetypes of Nature, Art, and ScienceDari EverandA Beginner's Guide to Constructing the Universe: The Mathematical Archetypes of Nature, Art, and SciencePenilaian: 4 dari 5 bintang4/5 (51)
- Midnight in Chernobyl: The Story of the World's Greatest Nuclear DisasterDari EverandMidnight in Chernobyl: The Story of the World's Greatest Nuclear DisasterPenilaian: 4.5 dari 5 bintang4.5/5 (410)
- Quantum Spirituality: Science, Gnostic Mysticism, and Connecting with Source ConsciousnessDari EverandQuantum Spirituality: Science, Gnostic Mysticism, and Connecting with Source ConsciousnessPenilaian: 4 dari 5 bintang4/5 (6)
- Lost in Math: How Beauty Leads Physics AstrayDari EverandLost in Math: How Beauty Leads Physics AstrayPenilaian: 4.5 dari 5 bintang4.5/5 (125)
- The End of Everything: (Astrophysically Speaking)Dari EverandThe End of Everything: (Astrophysically Speaking)Penilaian: 4.5 dari 5 bintang4.5/5 (157)
- Bedeviled: A Shadow History of Demons in ScienceDari EverandBedeviled: A Shadow History of Demons in SciencePenilaian: 5 dari 5 bintang5/5 (5)
- Let There Be Light: Physics, Philosophy & the Dimensional Structure of ConsciousnessDari EverandLet There Be Light: Physics, Philosophy & the Dimensional Structure of ConsciousnessPenilaian: 4.5 dari 5 bintang4.5/5 (57)
- The Power of Eight: Harnessing the Miraculous Energies of a Small Group to Heal Others, Your Life, and the WorldDari EverandThe Power of Eight: Harnessing the Miraculous Energies of a Small Group to Heal Others, Your Life, and the WorldPenilaian: 4.5 dari 5 bintang4.5/5 (54)
- The 60 Minute Quantum Physics Book: Science Made Easy For Beginners Without Math And In Plain Simple EnglishDari EverandThe 60 Minute Quantum Physics Book: Science Made Easy For Beginners Without Math And In Plain Simple EnglishPenilaian: 4.5 dari 5 bintang4.5/5 (4)
- Quantum Physcis for Beginners: An Easy Guide for Discovering the Hidden Side of Reality One Speck at a TimeDari EverandQuantum Physcis for Beginners: An Easy Guide for Discovering the Hidden Side of Reality One Speck at a TimeBelum ada peringkat
- Vibration and Frequency: How to Get What You Want in LifeDari EverandVibration and Frequency: How to Get What You Want in LifePenilaian: 4.5 dari 5 bintang4.5/5 (13)
- The Beginning of Infinity: Explanations That Transform the WorldDari EverandThe Beginning of Infinity: Explanations That Transform the WorldPenilaian: 5 dari 5 bintang5/5 (60)
- Quantum Physics: What Everyone Needs to KnowDari EverandQuantum Physics: What Everyone Needs to KnowPenilaian: 4.5 dari 5 bintang4.5/5 (49)
- A Natural History of Color: The Science Behind What We See and How We See itDari EverandA Natural History of Color: The Science Behind What We See and How We See itPenilaian: 4 dari 5 bintang4/5 (13)
- Beyond Weird: Why Everything You Thought You Knew about Quantum Physics Is DifferentDari EverandBeyond Weird: Why Everything You Thought You Knew about Quantum Physics Is DifferentPenilaian: 4 dari 5 bintang4/5 (25)
- Hyperspace: A Scientific Odyssey Through Parallel Universes, Time Warps, and the 10th DimensionDari EverandHyperspace: A Scientific Odyssey Through Parallel Universes, Time Warps, and the 10th DimensionPenilaian: 4.5 dari 5 bintang4.5/5 (3)
- The Holographic Universe: The Revolutionary Theory of RealityDari EverandThe Holographic Universe: The Revolutionary Theory of RealityPenilaian: 4.5 dari 5 bintang4.5/5 (78)