Anda mungkin juga menyukai
- CEEM PamfletDokumen2 halamanCEEM Pamfletapi-3705495Belum ada peringkat
- PsiquiatrÍa CDokumen1 halamanPsiquiatrÍa Capi-3705495Belum ada peringkat
- Reuma Primera VueltaDokumen179 halamanReuma Primera Vueltaapi-3705495100% (1)
- Derma Segunda VueltaDokumen153 halamanDerma Segunda Vueltaapi-3705495Belum ada peringkat
- Endocrino Segunda VueltaDokumen68 halamanEndocrino Segunda Vueltaapi-3705495Belum ada peringkat
- Manifestación Día 29 Abril, 12Dokumen8 halamanManifestación Día 29 Abril, 12api-3705495Belum ada peringkat
- Derma (Grupo II)Dokumen3 halamanDerma (Grupo II)api-3705495Belum ada peringkat
- PsiquiatrÍa DDokumen1 halamanPsiquiatrÍa Dapi-3705495Belum ada peringkat
- PsiquiatrÍa BDokumen1 halamanPsiquiatrÍa Bapi-3705495Belum ada peringkat
- LAS CUATRO TORRES (Modo de AdDokumen21 halamanLAS CUATRO TORRES (Modo de Adapi-3705495Belum ada peringkat
- Propuesta de Prácticas para Hospital RosellDokumen16 halamanPropuesta de Prácticas para Hospital Rosellapi-3705495Belum ada peringkat
- Médica DDokumen1 halamanMédica Dapi-37054950% (1)
- Cuaderno Practicas MédicaDokumen26 halamanCuaderno Practicas Médicaapi-3705495100% (2)
- Ficha de Practicas0708Dokumen2 halamanFicha de Practicas0708api-3698281Belum ada peringkat
- Farma CDokumen1 halamanFarma Capi-3705495Belum ada peringkat
- CIRUGIASDokumen4 halamanCIRUGIASapi-3705495Belum ada peringkat
- PsiquiatrÍa ADokumen1 halamanPsiquiatrÍa Aapi-3705495Belum ada peringkat
- Farma BDokumen1 halamanFarma Bapi-3705495Belum ada peringkat
- Farma DDokumen1 halamanFarma Dapi-3705495Belum ada peringkat
- Medica BDokumen1 halamanMedica Bapi-3705495Belum ada peringkat
- Medica CDokumen1 halamanMedica Capi-3705495Belum ada peringkat
- Farma ADokumen1 halamanFarma Aapi-3705495Belum ada peringkat
- Extra 25: Agresion Sexual - 2Dokumen2 halamanExtra 25: Agresion Sexual - 2api-3705495Belum ada peringkat
- Médica - ADokumen1 halamanMédica - Aapi-3705495Belum ada peringkat
- Urgencias y Emergencias IDokumen148 halamanUrgencias y Emergencias Iedgardojose100% (2)
- Extra 19: Intoxicaciones Por Metanol y EtilenglicolDokumen9 halamanExtra 19: Intoxicaciones Por Metanol y Etilenglicolapi-3705495100% (1)
- El M+®todo PericialDokumen6 halamanEl M+®todo Pericialapi-3705495100% (1)
- 10 11 2007 Toxicidad Cl+¡nicaDokumen13 halaman10 11 2007 Toxicidad Cl+¡nicaapi-3705495Belum ada peringkat
- Artículo 4 (Quedaba El Último Punto)Dokumen9 halamanArtículo 4 (Quedaba El Último Punto)api-3705495Belum ada peringkat
- Seminarios Medicina Legal - SeleneDokumen6 halamanSeminarios Medicina Legal - Seleneapi-3705495Belum ada peringkat
- Trabajo Reino AnimaliaDokumen12 halamanTrabajo Reino AnimaliaYefimovich GrigoriBelum ada peringkat
- Objetiv OsDokumen8 halamanObjetiv OsJassmin SantinBelum ada peringkat
- Qué Son Los AminoácidosDokumen36 halamanQué Son Los Aminoácidosana esperanza guerreroBelum ada peringkat
- Animales Vertebrados e Invertebrados, DG 2014Dokumen10 halamanAnimales Vertebrados e Invertebrados, DG 2014David GonzálezBelum ada peringkat
- Leuco GramaDokumen66 halamanLeuco GramaFranz HuamaniBelum ada peringkat
- Tarea - Metabolismo de Aminoacidos - Moscol Araujo, JimmyDokumen6 halamanTarea - Metabolismo de Aminoacidos - Moscol Araujo, JimmyFabricio Ronceros GregorioBelum ada peringkat
- Tensoactivos en FabricasDokumen6 halamanTensoactivos en FabricasLuis Fernando Ruiz DulantoBelum ada peringkat
- MontacerdosDokumen18 halamanMontacerdosRomina Meyer83% (6)
- Ilovepdf MergedDokumen81 halamanIlovepdf MergedSILVIA ROSARIO CHALCO MENDOZABelum ada peringkat
- Sistemas de FermentaciónDokumen30 halamanSistemas de FermentaciónEdwinCalderonMora100% (1)
- Celula VegetalDokumen8 halamanCelula VegetalNico SosaBelum ada peringkat
- Tonsiolitos (Puntos Blancos en La GargantaDokumen2 halamanTonsiolitos (Puntos Blancos en La GargantaCristian VidalBelum ada peringkat
- Guía de GenitourinarioDokumen18 halamanGuía de GenitourinarioMaria Fernanda Hernandez CastañedaBelum ada peringkat
- 2016-III Simulacro 06.03 CompletoDokumen11 halaman2016-III Simulacro 06.03 CompletoRoger Moreyra QuinchoBelum ada peringkat
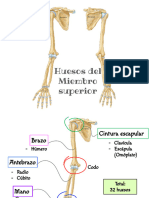
- Osteología Del Miembro SuperiorDokumen75 halamanOsteología Del Miembro SuperiorAna Carolina GaspariBelum ada peringkat
- Características de Un DiluyenteDokumen3 halamanCaracterísticas de Un DiluyenteluisfranvillalbaBelum ada peringkat
- Cortex Challenge - Braintopia - Branitopia Beyond Rulebook MultilingualDokumen20 halamanCortex Challenge - Braintopia - Branitopia Beyond Rulebook MultilingualAndrés EnriqueBelum ada peringkat
- Casos Clinicos 3Dokumen5 halamanCasos Clinicos 3capirote73Belum ada peringkat
- Analisis de HematocritoDokumen10 halamanAnalisis de HematocritoLuu MelendezBelum ada peringkat
- Actividad Sopa de LetrasDokumen2 halamanActividad Sopa de LetrasiTzArturoMC [PvP]Belum ada peringkat
- HONGOS (Modo de Compatibilidad)Dokumen63 halamanHONGOS (Modo de Compatibilidad)Emanuel PomaccoBelum ada peringkat
- 11.1 Oseo 2Dokumen85 halaman11.1 Oseo 2Nicole RodriguezBelum ada peringkat
- Ejemplo Cálculo de Protecciones de Alimentador en 13.8 KVDokumen3 halamanEjemplo Cálculo de Protecciones de Alimentador en 13.8 KVcluisyBelum ada peringkat
- Informe Grupo1 Zoolgia de InvertebradosDokumen43 halamanInforme Grupo1 Zoolgia de InvertebradosJunior CastilloBelum ada peringkat
- Tesis Flexibilidad CognitivaDokumen195 halamanTesis Flexibilidad CognitivaLourdes FloresBelum ada peringkat
- CUADRO COMPARATIVO SOBRE LA ESTRUCTURA Y FUNCIÓN de Los NeurotrasmisoresDokumen3 halamanCUADRO COMPARATIVO SOBRE LA ESTRUCTURA Y FUNCIÓN de Los Neurotrasmisoresjhazmin PeñaBelum ada peringkat
- Barreramac 101016000304 Phpapp02Dokumen39 halamanBarreramac 101016000304 Phpapp02Yhubi L. Beltran RuizBelum ada peringkat
- Plan FuncionalDokumen5 halamanPlan FuncionalMonica BenavidesBelum ada peringkat
- Pruebas de Función Hepática y GastrointestinalDokumen12 halamanPruebas de Función Hepática y GastrointestinalAlejandra LópezBelum ada peringkat
- Rhincodon TypusDokumen6 halamanRhincodon TypusbyrucBelum ada peringkat