Anda mungkin juga menyukai
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (119)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (587)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2219)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- Ssp393 - Audi A5 - Convenience Electronics and Driver Assist SystemsDokumen56 halamanSsp393 - Audi A5 - Convenience Electronics and Driver Assist SystemsCyberemuleBelum ada peringkat
- c5g Controller UnitDokumen254 halamanc5g Controller UnitElyBelum ada peringkat
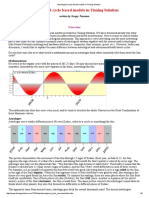
- Astrological Cycle Based Models in Timing Solution PDFDokumen9 halamanAstrological Cycle Based Models in Timing Solution PDFanalystbank100% (2)
- HDS Storage Foundations Exam Questions and AnswersDokumen42 halamanHDS Storage Foundations Exam Questions and Answersthuralwin85Belum ada peringkat
- JFET AC Equivalent CircuitsDokumen42 halamanJFET AC Equivalent Circuitssakuntala barikBelum ada peringkat
- DX5 EuserguideDokumen20 halamanDX5 Euserguidegabriel ferreyraBelum ada peringkat
- Ibm Websphere Application Server Advanced Edition, v3.5 - Websphere Jms - Jta Support For Mqseries (370K) WasDokumen49 halamanIbm Websphere Application Server Advanced Edition, v3.5 - Websphere Jms - Jta Support For Mqseries (370K) Wasimene IBBelum ada peringkat
- Face RecogDokumen26 halamanFace RecogFavasBelum ada peringkat
- Conectividad para Sistemas Industriales Jun 2018Dokumen49 halamanConectividad para Sistemas Industriales Jun 2018Cristian TorresBelum ada peringkat
- SyllabusDokumen2 halamanSyllabusBhavana NagarajBelum ada peringkat
- FieryOption Supported FierysDokumen4 halamanFieryOption Supported Fierysf559074Belum ada peringkat
- Maximabook PDFDokumen121 halamanMaximabook PDFWarley GramachoBelum ada peringkat
- Solving LCM HCF Remainder ProblemsDokumen10 halamanSolving LCM HCF Remainder ProblemshadhaiBelum ada peringkat
- Eiki LC-XT2 SMDokumen100 halamanEiki LC-XT2 SMgreggles777Belum ada peringkat
- MCQ - Unit 6 AutomationDokumen3 halamanMCQ - Unit 6 AutomationDipak naikBelum ada peringkat
- Biometrics 1Dokumen26 halamanBiometrics 1Naman JainBelum ada peringkat
- CCFL Inverter ZXDokumen26 halamanCCFL Inverter ZXcuzquilloBelum ada peringkat
- Manual de Configuración y Comisionamiento OptiX RTN - U2000 PDFDokumen40 halamanManual de Configuración y Comisionamiento OptiX RTN - U2000 PDFJonathan Gonzalez.LBelum ada peringkat
- Recruitment and Selection Centre: RSC Vacancy Bulletin No. 1/2013 Public Service Staff PostsDokumen48 halamanRecruitment and Selection Centre: RSC Vacancy Bulletin No. 1/2013 Public Service Staff PostsRich bitchBelum ada peringkat
- Spring CoreDokumen480 halamanSpring CoreANUSHA S GBelum ada peringkat
- Mock - 3sDokumen20 halamanMock - 3sPichuRangerBelum ada peringkat
- International Technology Roadmap For Semiconductors: 2008 Itrs OrtcDokumen23 halamanInternational Technology Roadmap For Semiconductors: 2008 Itrs Ortcvishal garadBelum ada peringkat
- How To Recover MSSQL From Suspect Mode Emergency Mode Error 1813Dokumen3 halamanHow To Recover MSSQL From Suspect Mode Emergency Mode Error 1813Scott NeubauerBelum ada peringkat
- ABB Price Book 714Dokumen1 halamanABB Price Book 714EliasBelum ada peringkat
- CS121Dokumen2 halamanCS121Ikon TrashBelum ada peringkat
- Reliance Jio Internship Report on Employee MotivationDokumen51 halamanReliance Jio Internship Report on Employee MotivationSahas ShettyBelum ada peringkat
- Contoh CVDokumen2 halamanContoh CVBetty Pasaribu100% (1)
- Nina Harris - Popper Lab Report 1Dokumen3 halamanNina Harris - Popper Lab Report 1api-647760982Belum ada peringkat
- Ipv4 Address Classes PDFDokumen2 halamanIpv4 Address Classes PDFAmol IngleBelum ada peringkat
- Control System Engineering: The LecturerDokumen8 halamanControl System Engineering: The LecturerХанифъ РахманBelum ada peringkat