Anda mungkin juga menyukai
- Hypothesis Tests For Principal Component Analysis When Variables Are StandardizedDokumen20 halamanHypothesis Tests For Principal Component Analysis When Variables Are StandardizedNikhilBelum ada peringkat
- GOVOROV M. (1), GIENKO G. (2) (1) Vancouver Island University, NANAIMO, CANADA (2) University of Alaska Anchorage, Anchorage, United StatesDokumen13 halamanGOVOROV M. (1), GIENKO G. (2) (1) Vancouver Island University, NANAIMO, CANADA (2) University of Alaska Anchorage, Anchorage, United StatesradocajdorijanBelum ada peringkat
- Overdispersion Models and EstimationDokumen20 halamanOverdispersion Models and EstimationJohann Sebastian ClaveriaBelum ada peringkat
- Some Methods For Analyzing and Correcting For Spatial AutocorrelationDokumen26 halamanSome Methods For Analyzing and Correcting For Spatial AutocorrelationFrancisco Parada SrepelBelum ada peringkat
- Beale Et Al 2010 Regression Analysis of Spatial DataDokumen19 halamanBeale Et Al 2010 Regression Analysis of Spatial DataValdemir SousaBelum ada peringkat
- Sizer Analysis For The Comparison of Regression Curves: Cheolwoo Park and Kee-Hoon KangDokumen27 halamanSizer Analysis For The Comparison of Regression Curves: Cheolwoo Park and Kee-Hoon KangjofrajodiBelum ada peringkat
- Stassopoulou 98Dokumen40 halamanStassopoulou 98Rony TeguhBelum ada peringkat
- XXX Report Version Oct 2012Dokumen38 halamanXXX Report Version Oct 2012zarcone7Belum ada peringkat
- Regional Flood Freq-1 Estimation Heo Et AlDokumen14 halamanRegional Flood Freq-1 Estimation Heo Et Alabejorro13Belum ada peringkat
- tmp8FC3 TMPDokumen6 halamantmp8FC3 TMPFrontiersBelum ada peringkat
- Use of CCA and CADokumen24 halamanUse of CCA and CASanet Janse van VuurenBelum ada peringkat
- Does Model-Free Forecasting Really Outperform The True Model?Dokumen5 halamanDoes Model-Free Forecasting Really Outperform The True Model?Peer TerBelum ada peringkat
- Scalable Variational Inference For BayesianDokumen36 halamanScalable Variational Inference For BayesianClaudio MelloBelum ada peringkat
- 19 Aos1924Dokumen22 halaman19 Aos1924sherlockholmes108Belum ada peringkat
- Maximum Likelihood Estimation of Limited and Discrete Dependent Variable Models With Nested Random EffectsDokumen23 halamanMaximum Likelihood Estimation of Limited and Discrete Dependent Variable Models With Nested Random EffectsJaime NavarroBelum ada peringkat
- Gibbs Sampler Approach For Objective Bayeisan Inference in Elliptical Multivariate Random Effects ModelDokumen32 halamanGibbs Sampler Approach For Objective Bayeisan Inference in Elliptical Multivariate Random Effects ModelChet MehtaBelum ada peringkat
- Modeling Spatial Uncertainty For Locally Uncertain DataDokumen12 halamanModeling Spatial Uncertainty For Locally Uncertain Dataseyyed81Belum ada peringkat
- Geostatistical Mixed Beta Regression A Bayesian ApproachDokumen14 halamanGeostatistical Mixed Beta Regression A Bayesian Approachamera naBelum ada peringkat
- Modeling of Species Distributions With Maxent: New Extensions and A Comprehensive EvaluationDokumen15 halamanModeling of Species Distributions With Maxent: New Extensions and A Comprehensive EvaluationDavid AarónBelum ada peringkat
- Application of A Bayesian Network in A GIS Based Decision Making SystemDokumen40 halamanApplication of A Bayesian Network in A GIS Based Decision Making SystemSyarifah NuzulBelum ada peringkat
- Parameter and Variance Estimation in Geotechnical Backanalysis Itsing Prior InformationDokumen23 halamanParameter and Variance Estimation in Geotechnical Backanalysis Itsing Prior InformationHUGIBelum ada peringkat
- Statistical Applications in Genetics and Molecular BiologyDokumen28 halamanStatistical Applications in Genetics and Molecular BiologyVirna DrakouBelum ada peringkat
- Stochastic Optimization: A Review: Dimitris Fouskakis and David DraperDokumen35 halamanStochastic Optimization: A Review: Dimitris Fouskakis and David DrapermuralimoviesBelum ada peringkat
- Final DeGeuser Lefebvre MRT2011Dokumen7 halamanFinal DeGeuser Lefebvre MRT2011Mert OvnBelum ada peringkat
- Mixture Models For Rating Data: The Method of Moments Via GR Obner BasisDokumen28 halamanMixture Models For Rating Data: The Method of Moments Via GR Obner BasisShijie ShengBelum ada peringkat
- Do Not Log-Transform Count DataDokumen5 halamanDo Not Log-Transform Count DataarrudajefersonBelum ada peringkat
- Comparing The Areas Under Two or More Correlated Receiver Operating Characteristic Curves A Nonparametric ApproachDokumen10 halamanComparing The Areas Under Two or More Correlated Receiver Operating Characteristic Curves A Nonparametric Approachajax_telamonioBelum ada peringkat
- Smooth Splines Large DataDokumen22 halamanSmooth Splines Large DataergoshaunBelum ada peringkat
- Compression Ratios Based On The Universal Similarity Metric Still Yield Protein Distances Far From CATH DistancesDokumen11 halamanCompression Ratios Based On The Universal Similarity Metric Still Yield Protein Distances Far From CATH DistancesPetra VitezBelum ada peringkat
- Background and Threshold: Critical Comparison of Methods of DeterminationDokumen16 halamanBackground and Threshold: Critical Comparison of Methods of DeterminationSoleh SundavaBelum ada peringkat
- Debinfer: Bayesian Inference For Dynamical Models of Biological Systems in RDokumen27 halamanDebinfer: Bayesian Inference For Dynamical Models of Biological Systems in RshidaoBelum ada peringkat
- Maruo Etal 2017Dokumen15 halamanMaruo Etal 2017Andre Luiz GrionBelum ada peringkat
- Graphical Displays For Meta-AnalysisDokumen15 halamanGraphical Displays For Meta-Analysisdinh son myBelum ada peringkat
- Geographically Weighted Regression Modelling Spatial NonstationarityDokumen14 halamanGeographically Weighted Regression Modelling Spatial NonstationarityDanang AriyantoBelum ada peringkat
- 2001, Pena, PrietoDokumen25 halaman2001, Pena, Prietomatchman6Belum ada peringkat
- Test of Homogeneity Based On Geometric Mean of VariancesDokumen11 halamanTest of Homogeneity Based On Geometric Mean of VariancesGlobal Research and Development ServicesBelum ada peringkat
- Legendre Fortin MER 2010Dokumen14 halamanLegendre Fortin MER 2010Catalina Verdugo ArriagadaBelum ada peringkat
- Principal Component Analysis For Data Containing Outliers and Missing ElementsDokumen16 halamanPrincipal Component Analysis For Data Containing Outliers and Missing ElementsDeeptimayee SahooBelum ada peringkat
- Eubank 2004Dokumen15 halamanEubank 2004Aurora LuckyBelum ada peringkat
- Cortical Correspondence With Probabilistic Fiber ConnectivityDokumen12 halamanCortical Correspondence With Probabilistic Fiber ConnectivitymarcniethammerBelum ada peringkat
- A New Method of Central Difference InterpolationDokumen14 halamanA New Method of Central Difference InterpolationAnonymous wGbPz3YBelum ada peringkat
- InTech-Data Association Techniques For Non Gaussian MeasurementsDokumen24 halamanInTech-Data Association Techniques For Non Gaussian MeasurementshallodriBelum ada peringkat
- Conjugate PriorDokumen47 halamanConjugate PriorShiyu XuBelum ada peringkat
- Tmpa153 TMPDokumen2 halamanTmpa153 TMPFrontiersBelum ada peringkat
- Ecography - 2007 - F Dormann - Methods To Account For Spatial Autocorrelation in The Analysis of Species DistributionalDokumen20 halamanEcography - 2007 - F Dormann - Methods To Account For Spatial Autocorrelation in The Analysis of Species DistributionalNicolas RuizBelum ada peringkat
- A Flexible Bayesian Variable Selection Approach For Modeling Interval DataDokumen20 halamanA Flexible Bayesian Variable Selection Approach For Modeling Interval DataSwapnaneel BhattacharyyaBelum ada peringkat
- Bickel and Levina 2004Dokumen28 halamanBickel and Levina 2004dfaini12Belum ada peringkat
- Comparison - Two Biomech Measure SystemsDokumen36 halamanComparison - Two Biomech Measure SystemsmtwakadBelum ada peringkat
- Clementi Diffussion MAps JCPDokumen11 halamanClementi Diffussion MAps JCPDavid BeveridgeBelum ada peringkat
- Bayesian Statistics For Small Area EstimationDokumen36 halamanBayesian Statistics For Small Area Estimationhasniii babaBelum ada peringkat
- Application of Copulas in Geostatistics: Claus P. Haslauer, Jing Li, and Andr As B ArdossyDokumen10 halamanApplication of Copulas in Geostatistics: Claus P. Haslauer, Jing Li, and Andr As B Ardossyseyyed81Belum ada peringkat
- Novkaniza 2019 IOP Conf. Ser. Earth Environ. Sci. 299 012030Dokumen14 halamanNovkaniza 2019 IOP Conf. Ser. Earth Environ. Sci. 299 012030Melinda SariBelum ada peringkat
- Exploring Matrix Generation Strategies in Isogeometric AnalysisDokumen20 halamanExploring Matrix Generation Strategies in Isogeometric AnalysisJorge Luis Garcia ZuñigaBelum ada peringkat
- A Family of Geographically Weighted Regression ModelsDokumen36 halamanA Family of Geographically Weighted Regression ModelsYantoSeptianBelum ada peringkat
- Partial Least Squares Discriminant Analysis: A Dimensionality Reduction Method To Classify Hyperspectral DataDokumen21 halamanPartial Least Squares Discriminant Analysis: A Dimensionality Reduction Method To Classify Hyperspectral DataJoseBelum ada peringkat
- PpcaDokumen13 halamanPpcaMaxGKBelum ada peringkat
- An Order Statistics Based Method For ConDokumen16 halamanAn Order Statistics Based Method For ConSayed SalarBelum ada peringkat
- Introduction To Modeling Spatial Processes Using Geostatistical AnalystDokumen27 halamanIntroduction To Modeling Spatial Processes Using Geostatistical AnalystDavid Huamani UrpeBelum ada peringkat
- YIC Chapter 1 (2) MKTDokumen63 halamanYIC Chapter 1 (2) MKTMebre WelduBelum ada peringkat
- Head Coverings BookDokumen86 halamanHead Coverings BookRichu RosarioBelum ada peringkat
- How To Configure PowerMACS 4000 As A PROFINET IO Slave With Siemens S7Dokumen20 halamanHow To Configure PowerMACS 4000 As A PROFINET IO Slave With Siemens S7kukaBelum ada peringkat
- ISO 27001 Introduction Course (05 IT01)Dokumen56 halamanISO 27001 Introduction Course (05 IT01)Sheik MohaideenBelum ada peringkat
- LP For EarthquakeDokumen6 halamanLP For Earthquakejelena jorgeoBelum ada peringkat
- Prevention of Waterborne DiseasesDokumen2 halamanPrevention of Waterborne DiseasesRixin JamtshoBelum ada peringkat
- PDFDokumen10 halamanPDFerbariumBelum ada peringkat
- Continue Practice Exam Test Questions Part 1 of The SeriesDokumen7 halamanContinue Practice Exam Test Questions Part 1 of The SeriesKenn Earl Bringino VillanuevaBelum ada peringkat
- Fundamentals of Public Health ManagementDokumen3 halamanFundamentals of Public Health ManagementHPMA globalBelum ada peringkat
- Thesis PaperDokumen53 halamanThesis PaperAnonymous AOOrehGZAS100% (1)
- Dwnload Full Principles of Economics 7th Edition Frank Solutions Manual PDFDokumen35 halamanDwnload Full Principles of Economics 7th Edition Frank Solutions Manual PDFmirthafoucault100% (8)
- Apron CapacityDokumen10 halamanApron CapacityMuchammad Ulil AidiBelum ada peringkat
- Eloy-Stock English Full PDFDokumen0 halamanEloy-Stock English Full PDFR.s. WartsBelum ada peringkat
- Applications SeawaterDokumen23 halamanApplications SeawaterQatar home RentBelum ada peringkat
- Acer N300 ManualDokumen50 halamanAcer N300 Manualc_formatBelum ada peringkat
- WarringFleets Complete PDFDokumen26 halamanWarringFleets Complete PDFlingshu8100% (1)
- Progressive Muscle RelaxationDokumen4 halamanProgressive Muscle RelaxationEstéphany Rodrigues ZanonatoBelum ada peringkat
- 8051 NotesDokumen61 halaman8051 Notessubramanyam62Belum ada peringkat
- SASS Prelims 2017 4E5N ADokumen9 halamanSASS Prelims 2017 4E5N ADamien SeowBelum ada peringkat
- Friction: Ultiple Hoice UestionsDokumen5 halamanFriction: Ultiple Hoice Uestionspk2varmaBelum ada peringkat
- Canon Powershot S50 Repair Manual (CHAPTER 4. PARTS CATALOG) PDFDokumen13 halamanCanon Powershot S50 Repair Manual (CHAPTER 4. PARTS CATALOG) PDFRita CaselliBelum ada peringkat
- Man As God Created Him, ThemDokumen3 halamanMan As God Created Him, ThemBOEN YATORBelum ada peringkat
- Scholastica: Mock 1Dokumen14 halamanScholastica: Mock 1Fatema KhatunBelum ada peringkat
- 7TH Maths F.a-1Dokumen1 halaman7TH Maths F.a-1Marrivada SuryanarayanaBelum ada peringkat
- Fire Protection in BuildingsDokumen2 halamanFire Protection in BuildingsJames Carl AriesBelum ada peringkat
- Evolution Army 3 R DadDokumen341 halamanEvolution Army 3 R DadStanisław DisęBelum ada peringkat
- Impact of Pantawid Pamilyang Pilipino Program On EducationDokumen10 halamanImpact of Pantawid Pamilyang Pilipino Program On EducationEllyssa Erika MabayagBelum ada peringkat
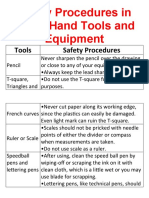
- Safety Procedures in Using Hand Tools and EquipmentDokumen12 halamanSafety Procedures in Using Hand Tools and EquipmentJan IcejimenezBelum ada peringkat
- Sources of Hindu LawDokumen9 halamanSources of Hindu LawKrishnaKousikiBelum ada peringkat
- Latched, Flip-Flops, and TimersDokumen36 halamanLatched, Flip-Flops, and TimersMuhammad Umair AslamBelum ada peringkat