Anda mungkin juga menyukai
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDari EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryPenilaian: 3.5 dari 5 bintang3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Dari EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Penilaian: 4.5 dari 5 bintang4.5/5 (121)
- Grit: The Power of Passion and PerseveranceDari EverandGrit: The Power of Passion and PerseverancePenilaian: 4 dari 5 bintang4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDari EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaPenilaian: 4.5 dari 5 bintang4.5/5 (266)
- The Little Book of Hygge: Danish Secrets to Happy LivingDari EverandThe Little Book of Hygge: Danish Secrets to Happy LivingPenilaian: 3.5 dari 5 bintang3.5/5 (400)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDari EverandNever Split the Difference: Negotiating As If Your Life Depended On ItPenilaian: 4.5 dari 5 bintang4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeDari EverandShoe Dog: A Memoir by the Creator of NikePenilaian: 4.5 dari 5 bintang4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerDari EverandThe Emperor of All Maladies: A Biography of CancerPenilaian: 4.5 dari 5 bintang4.5/5 (271)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDari EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifePenilaian: 4 dari 5 bintang4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDari EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyPenilaian: 3.5 dari 5 bintang3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDari EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersPenilaian: 4.5 dari 5 bintang4.5/5 (344)
- Rise of ISIS: A Threat We Can't IgnoreDari EverandRise of ISIS: A Threat We Can't IgnorePenilaian: 3.5 dari 5 bintang3.5/5 (137)
- Team of Rivals: The Political Genius of Abraham LincolnDari EverandTeam of Rivals: The Political Genius of Abraham LincolnPenilaian: 4.5 dari 5 bintang4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDari EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You ArePenilaian: 4 dari 5 bintang4/5 (1090)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDari EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RacePenilaian: 4 dari 5 bintang4/5 (895)
- Her Body and Other Parties: StoriesDari EverandHer Body and Other Parties: StoriesPenilaian: 4 dari 5 bintang4/5 (821)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDari EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FuturePenilaian: 4.5 dari 5 bintang4.5/5 (474)
- Kuan Yin Prayer Book.Dokumen11 halamanKuan Yin Prayer Book.AddySuBa85% (13)
- Plumbing Engineering Design Handbook, Vol 1 ASPEDokumen422 halamanPlumbing Engineering Design Handbook, Vol 1 ASPEMichelle NataliBelum ada peringkat
- The Unwinding: An Inner History of the New AmericaDari EverandThe Unwinding: An Inner History of the New AmericaPenilaian: 4 dari 5 bintang4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)Dari EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Penilaian: 4 dari 5 bintang4/5 (98)
- On Fire: The (Burning) Case for a Green New DealDari EverandOn Fire: The (Burning) Case for a Green New DealPenilaian: 4 dari 5 bintang4/5 (73)
- Carcinoma of Prostate: Dr. Saadat Hashmi Consultant UrologistDokumen48 halamanCarcinoma of Prostate: Dr. Saadat Hashmi Consultant UrologistMuhammad ArsalBelum ada peringkat
- Testbank 4e ch16Dokumen13 halamanTestbank 4e ch16drnazz100% (9)
- LEEDDokumen161 halamanLEEDjeruelBelum ada peringkat
- SinusitisDokumen402 halamanSinusitisIsidro Roberto Santana Gonzalez100% (1)
- 2 Day ALS Programme 24 Candidates IO ARS (March 2016) PDFDokumen2 halaman2 Day ALS Programme 24 Candidates IO ARS (March 2016) PDFCojocariu Emanuel50% (2)
- Sample Emg/Ncv Report - Normal StudyDokumen5 halamanSample Emg/Ncv Report - Normal StudyPhysiotherapist AliBelum ada peringkat
- 4 Strength and Durability of Concrete With LC3Dokumen41 halaman4 Strength and Durability of Concrete With LC3Mirza BasitBelum ada peringkat
- Cardiology ReviewDokumen12 halamanCardiology ReviewdrnazzBelum ada peringkat
- Biochem Q S Table ReviewDokumen27 halamanBiochem Q S Table ReviewMallick KodavantiBelum ada peringkat
- Reg GlycDokumen6 halamanReg GlycdrnazzBelum ada peringkat
- UW Sequence ExplanationsDokumen28 halamanUW Sequence ExplanationsdrnazzBelum ada peringkat
- Cardiology ReviewDokumen12 halamanCardiology ReviewdrnazzBelum ada peringkat
- 01GITDokumen50 halaman01GIThasankababiBelum ada peringkat
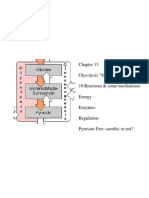
- Glycolysis "Sugar Splitting" 10 Reactions & Some Mechanisms Energy Enzymes Regulation Pyruvate Fate - Aerobic or Not?Dokumen45 halamanGlycolysis "Sugar Splitting" 10 Reactions & Some Mechanisms Energy Enzymes Regulation Pyruvate Fate - Aerobic or Not?drnazzBelum ada peringkat
- First Aid Extra NotesDokumen12 halamanFirst Aid Extra NotesdrnazzBelum ada peringkat
- Interruption of Bile Salt CirculationDokumen2 halamanInterruption of Bile Salt CirculationdrnazzBelum ada peringkat
- Sage Handbook For The USMLE Step 1 Class of 2013Dokumen17 halamanSage Handbook For The USMLE Step 1 Class of 2013drnazzBelum ada peringkat
- Interruption of Bile Salt CirculationDokumen2 halamanInterruption of Bile Salt CirculationdrnazzBelum ada peringkat
- GI Radiography: Astro Ntestinal EquenceDokumen7 halamanGI Radiography: Astro Ntestinal EquenceKath CacatianBelum ada peringkat
- 01GITDokumen50 halaman01GIThasankababiBelum ada peringkat
- OncogeneDokumen1 halamanOncogenedrnazzBelum ada peringkat
- Practice Acid-Base ProblemsDokumen3 halamanPractice Acid-Base ProblemsAmanda van TasselBelum ada peringkat
- Practice Acid-Base ProblemsDokumen3 halamanPractice Acid-Base ProblemsAmanda van TasselBelum ada peringkat
- Fuel EnergizerDokumen17 halamanFuel Energizerrakshak25100% (1)
- Steady State & Dynamic Wind Turbine ModelDokumen4 halamanSteady State & Dynamic Wind Turbine ModeldarshanraghuBelum ada peringkat
- Transmission Line BOQ VIMPDokumen72 halamanTransmission Line BOQ VIMPkajale_shrikant2325Belum ada peringkat
- Cleaning Disinfecting School ClassroomsDokumen2 halamanCleaning Disinfecting School ClassroomsFitz JaminitBelum ada peringkat
- Autodrill Satellite ManualDokumen37 halamanAutodrill Satellite ManualmiguelBelum ada peringkat
- How To Conduct A Situation AnalysisDokumen10 halamanHow To Conduct A Situation AnalysisÂmany AymanBelum ada peringkat
- vdYoyHdeTKeL7EhJwoXE - Insomnia PH SlidesDokumen40 halamanvdYoyHdeTKeL7EhJwoXE - Insomnia PH SlidesKreshnik IdrizajBelum ada peringkat
- 65 70Dokumen6 halaman65 70kang soon cheolBelum ada peringkat
- Black and Decker Vaporera Electrica RecetarioDokumen17 halamanBlack and Decker Vaporera Electrica RecetarioFabio AtenógenesBelum ada peringkat
- BFPPPDokumen15 halamanBFPPPFaith JacalanBelum ada peringkat
- Identification of Plastics Identification of PlasticsDokumen41 halamanIdentification of Plastics Identification of PlasticsSoumickBelum ada peringkat
- Measurement of Poverty and Poverty of Measurement: Martin GreeleyDokumen15 halamanMeasurement of Poverty and Poverty of Measurement: Martin GreeleyKule89Belum ada peringkat
- Effect of Different Immediate Dentin Sealing Techniques On The Microtensile Bond Strength PDFDokumen6 halamanEffect of Different Immediate Dentin Sealing Techniques On The Microtensile Bond Strength PDFclaudiaBelum ada peringkat
- New Microsoft Office Word DocumentDokumen3 halamanNew Microsoft Office Word DocumentSunija SelvamBelum ada peringkat
- 1109 KW 1,487 HP: Gross HorsepowerDokumen12 halaman1109 KW 1,487 HP: Gross HorsepowerDedek SukmaBelum ada peringkat
- Maxipro NewDokumen2 halamanMaxipro NewLokendraBelum ada peringkat
- 250 Watt Solar Panel SpecificationsDokumen2 halaman250 Watt Solar Panel Specificationsfopoku2k20% (1)
- Forensic Toxicology: A. Classify Toxins and Their Effects On The BodyDokumen28 halamanForensic Toxicology: A. Classify Toxins and Their Effects On The BodySajid RehmanBelum ada peringkat
- The Power of Partnership: Underground Room & Pillar Lateral Development and DownholesDokumen4 halamanThe Power of Partnership: Underground Room & Pillar Lateral Development and DownholesjoxegutierrezgBelum ada peringkat
- Morgan - Electrostatics 2003 (Institute of Physics Conference Series) - Institute of Physics Publishing (2004)Dokumen386 halamanMorgan - Electrostatics 2003 (Institute of Physics Conference Series) - Institute of Physics Publishing (2004)Tsiory RanaivosonBelum ada peringkat
- Comparatives and SuperlativesDokumen2 halamanComparatives and Superlativesjcarlosgf60% (5)
- Assessment of Reading Comprehension 2Dokumen8 halamanAssessment of Reading Comprehension 2Kutu DemangBelum ada peringkat