Anda mungkin juga menyukai
- Blast 2 Sequences, A New Tool For Comparing Protein and Nucleotide SequencesDokumen17 halamanBlast 2 Sequences, A New Tool For Comparing Protein and Nucleotide SequencesXâlmañ KháñBelum ada peringkat
- Pattern Recognition 1Dokumen5 halamanPattern Recognition 1Tejinder SinghBelum ada peringkat
- Sequence Analysis - AlignmentDokumen57 halamanSequence Analysis - Alignmentfilson.riyadiBelum ada peringkat
- GlOsario BioinformaticaDokumen5 halamanGlOsario BioinformaticacristianojppBelum ada peringkat
- Bioinfo Notes 2Dokumen9 halamanBioinfo Notes 2Raj LonkarBelum ada peringkat
- Need & Emergence of The Field: Speaker Shashi Shekhar Head of Computational Section Biowits Life SciencesDokumen59 halamanNeed & Emergence of The Field: Speaker Shashi Shekhar Head of Computational Section Biowits Life SciencesIma kuro-bikeBelum ada peringkat
- Multiple Sequence Alignment ThesisDokumen8 halamanMultiple Sequence Alignment Thesisvalerielohkamprochester100% (2)
- Lab Report 3 BioinformaticsDokumen18 halamanLab Report 3 BioinformaticsRabiatul Adawiyah HasbullahBelum ada peringkat
- Homologymodeling 150123025144 Conversion Gate01Dokumen27 halamanHomologymodeling 150123025144 Conversion Gate01Jhanvi SBelum ada peringkat
- Bioinformatics AnswersDokumen13 halamanBioinformatics AnswersPratibha Patil100% (1)
- BI ManualDokumen35 halamanBI ManualFazilaBelum ada peringkat
- Blast 2 Sequences: Salman Khan Current Gpa in Bioinf 4 GpaDokumen45 halamanBlast 2 Sequences: Salman Khan Current Gpa in Bioinf 4 GpaXâlmañ KháñBelum ada peringkat
- Structural BioinfoDokumen76 halamanStructural BioinfoMudit MisraBelum ada peringkat
- Bio in For Ma TicsDokumen54 halamanBio in For Ma TicsKaveesh DashoraBelum ada peringkat
- Thesis On Homology ModelingDokumen6 halamanThesis On Homology ModelingWriteMyPaperFastUK100% (2)
- PSI-BLAST Tutorial - Comparative Genomics-For Term PaperDokumen9 halamanPSI-BLAST Tutorial - Comparative Genomics-For Term PaperDevinder KaurBelum ada peringkat
- Difference between local and global alignmentDokumen6 halamanDifference between local and global alignmentGum Naam SingerBelum ada peringkat
- Sequence Alignment Methods and ApplicationsDokumen5 halamanSequence Alignment Methods and ApplicationsGopi ShankarBelum ada peringkat
- Lab Report 05Dokumen20 halamanLab Report 05DewBelum ada peringkat
- Current solved BIF 401 paperDokumen16 halamanCurrent solved BIF 401 paperSagheer MalikBelum ada peringkat
- BLASTDokumen4 halamanBLASTdia320576Belum ada peringkat
- UntitledDokumen13 halamanUntitledDewBelum ada peringkat
- H ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisDokumen10 halamanH ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisijcseitBelum ada peringkat
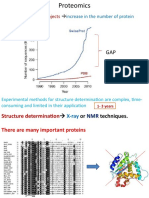
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDokumen27 halamanGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasBelum ada peringkat
- Lab WorkDokumen29 halamanLab WorkAleena KhanBelum ada peringkat
- BLAST Glossary With HighlightsDokumen9 halamanBLAST Glossary With Highlightsimran47Belum ada peringkat
- Lab Report 03Dokumen18 halamanLab Report 03DewBelum ada peringkat
- Unit III - P1Dokumen51 halamanUnit III - P1Suraj VermaBelum ada peringkat
- CCCCCCCCCCCCCCCCC C CCCCCCCCCCCCCCCCCCCCCDokumen42 halamanCCCCCCCCCCCCCCCCC C CCCCCCCCCCCCCCCCCCCCCAnkur PandeyBelum ada peringkat
- Basics of BioinformaticsDokumen99 halamanBasics of BioinformaticsSalam Pradeep Singh100% (5)
- Sequence AnalysisDokumen6 halamanSequence Analysisharrison9Belum ada peringkat
- Sequence Alignment: Sequence Alignment Is The Most Important Task in Bioinformatics!Dokumen13 halamanSequence Alignment: Sequence Alignment Is The Most Important Task in Bioinformatics!Malik FhaimBelum ada peringkat
- Bioinformatics by MHNDokumen15 halamanBioinformatics by MHNMuhammad Mubeen GahoBelum ada peringkat
- BIOINFORMATICSDokumen85 halamanBIOINFORMATICSlegendofrohithBelum ada peringkat
- Bif401 Solved Final Papers 2017Dokumen8 halamanBif401 Solved Final Papers 2017HRrehmanBelum ada peringkat
- Bioinfo Final PracticalDokumen66 halamanBioinfo Final PracticalPolu ChattopadhyayBelum ada peringkat
- Bio ModelDokumen12 halamanBio ModelDouuBelum ada peringkat
- Class Pairwise AlignmentDokumen17 halamanClass Pairwise AlignmentRoopak BeheraBelum ada peringkat
- Sequence Analysis 2Dokumen13 halamanSequence Analysis 2Tejinder SinghBelum ada peringkat
- Generation of Count Matrix - Introduction To Single-Cell RNA-seqDokumen9 halamanGeneration of Count Matrix - Introduction To Single-Cell RNA-seqYanelisa PulaniBelum ada peringkat
- BLAST Algorithm Practical Assignment Detect Frameshift Mutations Structurally Similar ProteinsDokumen5 halamanBLAST Algorithm Practical Assignment Detect Frameshift Mutations Structurally Similar Proteinslucy kabazziBelum ada peringkat
- Bioinformatics Tools: Stuart M. Brown, PH.D Dept of Cell Biology NYU School of MedicineDokumen50 halamanBioinformatics Tools: Stuart M. Brown, PH.D Dept of Cell Biology NYU School of MedicineVivek ChandelBelum ada peringkat
- Introduction To Bioinformatics: Tolga CanDokumen21 halamanIntroduction To Bioinformatics: Tolga CanvinothlcBelum ada peringkat
- Bioinformatics Pratical FileDokumen63 halamanBioinformatics Pratical FileSudheshnaBelum ada peringkat
- Introduction To Bioinformatics Lecture 3Dokumen20 halamanIntroduction To Bioinformatics Lecture 3HaMmAd KhAnBelum ada peringkat
- Application in Establishing Epidemiology and Variability: Genome & Protein " Sequence Analysis Programs"Dokumen23 halamanApplication in Establishing Epidemiology and Variability: Genome & Protein " Sequence Analysis Programs"Dr. Rajesh Kumar100% (3)
- Blast (Basic Local Alignment Search Tool)Dokumen28 halamanBlast (Basic Local Alignment Search Tool)yasasveBelum ada peringkat
- Bioinformatics Unit IDokumen6 halamanBioinformatics Unit IAditya SaxenaBelum ada peringkat
- Research Papers On Multiple Sequence AlignmentDokumen5 halamanResearch Papers On Multiple Sequence Alignmentafeartslf100% (1)
- Bioinformatics Tutorial 2016Dokumen28 halamanBioinformatics Tutorial 2016Rhie GieBelum ada peringkat
- Bio in For MaticsDokumen18 halamanBio in For Maticsowloabi ridwanBelum ada peringkat
- Chapter 2 BioinformaticsDokumen9 halamanChapter 2 BioinformaticsKaiash M YBelum ada peringkat
- Unit2 2Dokumen30 halamanUnit2 2sadhanaaBelum ada peringkat
- Bioinformatics (STH Sir)Dokumen13 halamanBioinformatics (STH Sir)Ranojoy SenBelum ada peringkat
- Lec2 BLAST Algorithm 2022Dokumen32 halamanLec2 BLAST Algorithm 2022Jak OkBelum ada peringkat
- 18bth028 Journal BioinformaticsDokumen13 halaman18bth028 Journal BioinformaticsLakshya PhoolwaniBelum ada peringkat
- Summary Bioinformation TechnologyDokumen15 halamanSummary Bioinformation TechnologytjBelum ada peringkat
- BlastDokumen6 halamanBlastGanduri VsrkBelum ada peringkat
- Introduction to Bioinformatics, Sequence and Genome AnalysisDari EverandIntroduction to Bioinformatics, Sequence and Genome AnalysisBelum ada peringkat
- Phylogenetic Trees (BIOINFORMATICS)Dokumen7 halamanPhylogenetic Trees (BIOINFORMATICS)Aayudh DasBelum ada peringkat
- Arish CVDokumen2 halamanArish CVArish QureshiBelum ada peringkat
- 7th 8th Syll With Objectivesa21.11.09acmDokumen36 halaman7th 8th Syll With Objectivesa21.11.09acmKiran KumarBelum ada peringkat
- Study Programme Details 3Dokumen2 halamanStudy Programme Details 3dcbdatBelum ada peringkat
- Statistics Genomics Quiz4Dokumen3 halamanStatistics Genomics Quiz4AbbasBelum ada peringkat
- H. pylori Primer SequencesDokumen3 halamanH. pylori Primer Sequencesasaleh74Belum ada peringkat
- Laboratory Manual: Biology 3055Dokumen37 halamanLaboratory Manual: Biology 3055Raj Kumar SoniBelum ada peringkat
- Top 50 Biotechnology Blogs - The BioBlogging ProjectDokumen5 halamanTop 50 Biotechnology Blogs - The BioBlogging ProjectDiana ElamBelum ada peringkat
- BiologicalsciencesDokumen10 halamanBiologicalsciencesapi-341408803Belum ada peringkat
- Bio PythonDokumen357 halamanBio PythonNiko Didic100% (1)
- UCSC Genome BrowserDokumen9 halamanUCSC Genome Browsernoah676Belum ada peringkat
- Examinition Cell BJBDokumen35 halamanExaminition Cell BJBSangram PandaBelum ada peringkat
- Microbial Detection Total Solution: MGI Sequencing Platform For Pathogen Fast IdentificationDokumen20 halamanMicrobial Detection Total Solution: MGI Sequencing Platform For Pathogen Fast IdentificationEntio BaezBelum ada peringkat
- Postdoc Position in David Chen Lab at City of HopeDokumen2 halamanPostdoc Position in David Chen Lab at City of HopeAdolfo De La Rosa JrBelum ada peringkat
- Presentation of BioinformaticsDokumen25 halamanPresentation of BioinformaticsMudassar SamarBelum ada peringkat
- Department of Computer ScienceDokumen2 halamanDepartment of Computer ScienceAnonymous XZZ6j8NOBelum ada peringkat
- Resume SsDokumen3 halamanResume SsShantanu ShuklaBelum ada peringkat
- Finalized Report 17nov2018 REVISEDDokumen30 halamanFinalized Report 17nov2018 REVISEDMario WallaceBelum ada peringkat
- Animais TransgênicosDokumen11 halamanAnimais TransgênicosCleidson OliveiraBelum ada peringkat
- Lecture 1: INTRODUCTION: A/Prof. Ly Le School of Biotechnology Email: Office: RM 705Dokumen43 halamanLecture 1: INTRODUCTION: A/Prof. Ly Le School of Biotechnology Email: Office: RM 705LinhNguyeBelum ada peringkat
- ResumeDokumen3 halamanResumeroopesh_cvBelum ada peringkat
- Bioinformatics: Analyzing DNA Sequence Using BLASTDokumen30 halamanBioinformatics: Analyzing DNA Sequence Using BLASTVinayak DoifodeBelum ada peringkat
- Master of Science IN Plant Molecular Biology and BiotechnologyDokumen33 halamanMaster of Science IN Plant Molecular Biology and BiotechnologyAryan KulhariBelum ada peringkat
- Bioinformatics Trends and Methodologies PDFDokumen736 halamanBioinformatics Trends and Methodologies PDFCrystal GranadosBelum ada peringkat
- Deep Learning in Bioinformatics: Seonwoo Min, Byunghan Lee and Sungroh YoonDokumen19 halamanDeep Learning in Bioinformatics: Seonwoo Min, Byunghan Lee and Sungroh YoonSyahrul Chotamy AZBelum ada peringkat
- Cerca de 1,910,000 Resultados (0.26 Segundos) : RGD - Mcw.eduDokumen2 halamanCerca de 1,910,000 Resultados (0.26 Segundos) : RGD - Mcw.edutlowdidj33Belum ada peringkat
- Edson Arantes Do NascimentoDokumen3 halamanEdson Arantes Do NascimentosmithaslBelum ada peringkat
- Cloud Computing FundamentalsDokumen31 halamanCloud Computing FundamentalsvizierBelum ada peringkat
- UF Microbiology Lab SyllabusDokumen22 halamanUF Microbiology Lab SyllabusAlessandra De SilvaBelum ada peringkat
- Human Genome Project and DNA FingerprintingDokumen2 halamanHuman Genome Project and DNA Fingerprintingnicholasmith80% (10)