Anda mungkin juga menyukai
- Nanoporous Catalysts for Biomass ConversionDari EverandNanoporous Catalysts for Biomass ConversionFeng-Shou XiaoBelum ada peringkat
- Chapter 23 Thermophilic Biohydrogen Production PDFDokumen12 halamanChapter 23 Thermophilic Biohydrogen Production PDFaegosmithBelum ada peringkat
- Advances in Biofeedstocks and Biofuels, Volume 2: Production Technologies for BiofuelsDari EverandAdvances in Biofeedstocks and Biofuels, Volume 2: Production Technologies for BiofuelsLalit Kumar SinghBelum ada peringkat
- Catalytic Formation of Lactic and Levulinic Acids From Biomass Derived Monosaccarides Through Sn-Beta Formed by ImpregnationDokumen16 halamanCatalytic Formation of Lactic and Levulinic Acids From Biomass Derived Monosaccarides Through Sn-Beta Formed by ImpregnationAhmed EssamBelum ada peringkat
- Processes 09 00436 v4Dokumen23 halamanProcesses 09 00436 v4Maheesha GunathungaBelum ada peringkat
- Using Waste CO To Increase Ethanol Production From Corn Ethanol Biorefineries: Techno-Economic AnalysisDokumen15 halamanUsing Waste CO To Increase Ethanol Production From Corn Ethanol Biorefineries: Techno-Economic Analysisnileshparmar21Belum ada peringkat
- Renewable HydrogenDokumen10 halamanRenewable HydrogenVedavathi ReddyBelum ada peringkat
- Green Chemistry: Accepted ManuscriptDokumen10 halamanGreen Chemistry: Accepted ManuscriptSyarif HidayatBelum ada peringkat
- Evaluation of Lignocellulosic Biomass Upgrading Routes To Fuels and ChemicalsDokumen21 halamanEvaluation of Lignocellulosic Biomass Upgrading Routes To Fuels and Chemicals김병철Belum ada peringkat
- An Integrated Catalytic Approach For The Production of Hydrogen by GlycerolDokumen6 halamanAn Integrated Catalytic Approach For The Production of Hydrogen by GlycerolMahdy HajienayatiBelum ada peringkat
- Sorption Enhanced Steam Reforming (SESR) : A Direct Route Towards Efficient Hydrogen Production From Biomass-Derived CompoundsDokumen8 halamanSorption Enhanced Steam Reforming (SESR) : A Direct Route Towards Efficient Hydrogen Production From Biomass-Derived CompoundsserchBelum ada peringkat
- PetroleumDokumen13 halamanPetroleumPrashantBelum ada peringkat
- Zhang 2015Dokumen7 halamanZhang 2015AishBelum ada peringkat
- Chemical Conversion of Biomass To Green ChemicalsDokumen32 halamanChemical Conversion of Biomass To Green ChemicalsMaria Cecille Sarmiento GarciaBelum ada peringkat
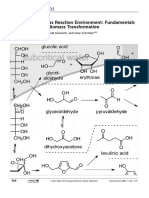
- Subcritical Water As Reaction Environment: Fundamentals of Hydrothermal Biomass TransformationDokumen14 halamanSubcritical Water As Reaction Environment: Fundamentals of Hydrothermal Biomass TransformationSeptian Perwira YudhaBelum ada peringkat
- Dnv-Position Paper Co2 Utilization Tcm4-445820Dokumen20 halamanDnv-Position Paper Co2 Utilization Tcm4-445820lhphong021191Belum ada peringkat
- Effect of Substrate Concentration On Hydrogen Production andDokumen9 halamanEffect of Substrate Concentration On Hydrogen Production andProfessor Douglas TorresBelum ada peringkat
- Simulation Studies On Ethanol Production From Sugar Cane ResiduesDokumen22 halamanSimulation Studies On Ethanol Production From Sugar Cane ResiduesChakuliBelum ada peringkat
- 1 s2.0 S0920586109000984 MainDokumen11 halaman1 s2.0 S0920586109000984 MainClaudia Elizabeth Ruiz DávilaBelum ada peringkat
- Reator Artigo PDFDokumen11 halamanReator Artigo PDFÊnio BruceBelum ada peringkat
- Ferment Able SugarDokumen6 halamanFerment Able SugarXenia MenaBelum ada peringkat
- Egeberg 2010 - Hydrotreating in The Production ofDokumen13 halamanEgeberg 2010 - Hydrotreating in The Production ofNadia RizanedewiBelum ada peringkat
- Energies 13 00610 v2 PDFDokumen25 halamanEnergies 13 00610 v2 PDFIsmail ŞahbazBelum ada peringkat
- Energies: /Zno/AlDokumen25 halamanEnergies: /Zno/AlAnonymous Ksq1dyPRhBelum ada peringkat
- ManuscriptDokumen18 halamanManuscriptNguyen TrangBelum ada peringkat
- Kinetics and Reaction Engineering of Levulinic Acid Production From Aqueous Glucose SolutionsDokumen12 halamanKinetics and Reaction Engineering of Levulinic Acid Production From Aqueous Glucose Solutionsראול אפונטהBelum ada peringkat
- Hydrogen ProductiomDokumen11 halamanHydrogen ProductiomKhoirul SaputriBelum ada peringkat
- A New Power, Methanol, and DME Polygeneration Process Using Integrated Chemical Looping SystemsDokumen15 halamanA New Power, Methanol, and DME Polygeneration Process Using Integrated Chemical Looping SystemsCriveanuNNarcisBelum ada peringkat
- Development of An Efficient Methanol Production PRDokumen31 halamanDevelopment of An Efficient Methanol Production PRKorean Drama TVBelum ada peringkat
- Methanol Production by CO Hydrogenation: Analysis and Simulation of Reactor PerformanceDokumen19 halamanMethanol Production by CO Hydrogenation: Analysis and Simulation of Reactor PerformancehelloBelum ada peringkat
- Hydrogen Production From Glycerol An UpdateDokumen5 halamanHydrogen Production From Glycerol An Updateemi_v11Belum ada peringkat
- Role of Photocatalysis - Recent AdvancementsDokumen20 halamanRole of Photocatalysis - Recent AdvancementsGRagaBelum ada peringkat
- Production of 5-HMF From Cellulosic Biomass: Experimental Results and Integrated Process SimulationDokumen13 halamanProduction of 5-HMF From Cellulosic Biomass: Experimental Results and Integrated Process SimulationCelic RamosBelum ada peringkat
- Producing Hydrogen Through Electrolysis and Other ProcessesDokumen50 halamanProducing Hydrogen Through Electrolysis and Other ProcessesAnedo 1909Belum ada peringkat
- Journal of CO Utilization: Nora Meiri, Roman Radus, Moti HerskowitzDokumen6 halamanJournal of CO Utilization: Nora Meiri, Roman Radus, Moti HerskowitzFarah Talib Al-sudaniBelum ada peringkat
- High PressureDokumen20 halamanHigh PressureJam imtiazBelum ada peringkat
- Python Project SynopsisDokumen30 halamanPython Project SynopsisAshish RoshanBelum ada peringkat
- Ismaila2021 Article ThermodynamicAnalysisOfSteamReDokumen12 halamanIsmaila2021 Article ThermodynamicAnalysisOfSteamRechairunnisaBelum ada peringkat
- SmolinskiHowaniec 16Dokumen11 halamanSmolinskiHowaniec 16Jesús Alfonso Vázquez BarragánBelum ada peringkat
- Carbon Capture: The Relevant Chemical Reactions For Hydrocarbon Fuel Production AreDokumen2 halamanCarbon Capture: The Relevant Chemical Reactions For Hydrocarbon Fuel Production AreWilliamBelum ada peringkat
- Solid Acid Catalyzed Aldol Dimerization of Levulinic Acid For The Preparation of C10 Renewable Fuel and Chemical FeedstocksDokumen6 halamanSolid Acid Catalyzed Aldol Dimerization of Levulinic Acid For The Preparation of C10 Renewable Fuel and Chemical FeedstocksLucas de MeloBelum ada peringkat
- s2.0 S0920586122X001141 s2.0 S0920586122000992main - PDFX Amz Security Token IQoJbDokumen18 halamans2.0 S0920586122X001141 s2.0 S0920586122000992main - PDFX Amz Security Token IQoJbMasroorAbroBelum ada peringkat
- 1 s2.0 S0360319922042458 MainDokumen11 halaman1 s2.0 S0360319922042458 Mainranjani093Belum ada peringkat
- Various Methods For Hydrogen Production - After EditDokumen11 halamanVarious Methods For Hydrogen Production - After EditmoftajBelum ada peringkat
- Energy Conversion and ManagementDokumen7 halamanEnergy Conversion and ManagementhusseinhshBelum ada peringkat
- Preproof HKirschDokumen51 halamanPreproof HKirschMohammed GhanemBelum ada peringkat
- Biogas Purification Using AmineDokumen9 halamanBiogas Purification Using AmineAlbertIvanoAndreanBelum ada peringkat
- Echno Economic Analysis For ProductionofLArabitol fromLArabinoseDokumen8 halamanEchno Economic Analysis For ProductionofLArabitol fromLArabinoseADITI AWASTHIBelum ada peringkat
- Materials 16 00472 v2Dokumen15 halamanMaterials 16 00472 v2Claudio Contreras DíazBelum ada peringkat
- Production of Renewable Hydrogen by Aqueous-Phase Reforming of Glycerol OverDokumen7 halamanProduction of Renewable Hydrogen by Aqueous-Phase Reforming of Glycerol OverMahdy HajienayatiBelum ada peringkat
- Sulfur and Hydrogen Sulfide RecoveryDokumen27 halamanSulfur and Hydrogen Sulfide RecoveryChemical.AliBelum ada peringkat
- ECM - Campanario - Gutierrez - 2017 - FT Biofuels From SCWR - ECM - PreprintDokumen40 halamanECM - Campanario - Gutierrez - 2017 - FT Biofuels From SCWR - ECM - PreprintAlbert StratingBelum ada peringkat
- Chemical Engineering Journal: Yunfei He, Luxin Zhang, Yuting Liu, Simin Yi, Han Yu, Yujie Zhu, Ruijun SunDokumen12 halamanChemical Engineering Journal: Yunfei He, Luxin Zhang, Yuting Liu, Simin Yi, Han Yu, Yujie Zhu, Ruijun Sunbruno barrosBelum ada peringkat
- Energies: Bio-Crude Production Through Aqueous Phase Recycling of Hydrothermal Liquefaction of Sewage SludgeDokumen18 halamanEnergies: Bio-Crude Production Through Aqueous Phase Recycling of Hydrothermal Liquefaction of Sewage SludgekrisBelum ada peringkat
- Selective Hydrolysis of Cellulose and Polysaccharides Into Sugars by Catalytic Hydrothermal Method Using Sulfonated Activated-CarbonDokumen14 halamanSelective Hydrolysis of Cellulose and Polysaccharides Into Sugars by Catalytic Hydrothermal Method Using Sulfonated Activated-CarbonFitri HardiantiBelum ada peringkat
- 3-D Multi-Tubular Reactor Model Development For The Oxidative Dehydrogenation of Butene To 1,3-ButadieneDokumen21 halaman3-D Multi-Tubular Reactor Model Development For The Oxidative Dehydrogenation of Butene To 1,3-ButadieneKhabib FirmansyahBelum ada peringkat
- New Horizons For Hydrogen: Producing Hydrogen From Renewable ResourcesDokumen8 halamanNew Horizons For Hydrogen: Producing Hydrogen From Renewable ResourcesBraulio BrunaudBelum ada peringkat
- Energies: Fractionation For Biodiesel Purification Using Supercritical Carbon DioxideDokumen10 halamanEnergies: Fractionation For Biodiesel Purification Using Supercritical Carbon DioxideArdila Hayu TiwikramaBelum ada peringkat
- BiohidrogenDokumen11 halamanBiohidrogenMinea Maria-ramonaBelum ada peringkat
- 2015-Anjos - Vitor - Diss-Use of Thermal Analysis To Control The Solidification MorphologyDokumen197 halaman2015-Anjos - Vitor - Diss-Use of Thermal Analysis To Control The Solidification MorphologyBahubali KabnureBelum ada peringkat
- Water TreatmentDokumen134 halamanWater TreatmentDangolBelum ada peringkat
- Basic Engineering ChemistryDokumen289 halamanBasic Engineering ChemistryNELLAIAPPAN SANKARAKUTTALAM73% (11)
- Aluminum: Eriochrome Cyanine R Method Method 8326 0.006 To 0.250 MG/L Al Powder PillowsDokumen8 halamanAluminum: Eriochrome Cyanine R Method Method 8326 0.006 To 0.250 MG/L Al Powder PillowsPedro MamaniBelum ada peringkat
- 2004 12 WJDokumen116 halaman2004 12 WJJoel CristobalBelum ada peringkat
- Activity 5 Chemical Reactions and Balancing Chemical Equations IDokumen6 halamanActivity 5 Chemical Reactions and Balancing Chemical Equations INivla Genesis100% (2)
- PHEngg Handouts and Workbook 4Dokumen175 halamanPHEngg Handouts and Workbook 4AMmarBelum ada peringkat
- ΔΤΦ 300413Dokumen2.180 halamanΔΤΦ 300413farmakopoioiBelum ada peringkat
- 6110 Aluminum Composition SpecDokumen1 halaman6110 Aluminum Composition SpecSung-Sil, JungBelum ada peringkat
- Nursing BundleDokumen107 halamanNursing BundleNina Klairea Padua60% (5)
- Atoms and MoleculesDokumen28 halamanAtoms and MoleculesthinkiitBelum ada peringkat
- Tarobiscend PDFDokumen12 halamanTarobiscend PDFGina AshfihaniBelum ada peringkat
- Salt WasheryDokumen7 halamanSalt WasheryABelum ada peringkat
- Topical Questions Form 1 - ChemistryDokumen37 halamanTopical Questions Form 1 - ChemistrySYLVIA CHEPKEMBOIBelum ada peringkat
- Janaushadhi Generic ListDokumen28 halamanJanaushadhi Generic ListSaqib Faheem KachrooBelum ada peringkat
- Experiment: Soil Chemical Analysis - Calcium CarbonateDokumen5 halamanExperiment: Soil Chemical Analysis - Calcium CarbonatedoraBelum ada peringkat
- Water Resistance of Basic Magnesium Sulfate Cement: SMAE 2016Dokumen9 halamanWater Resistance of Basic Magnesium Sulfate Cement: SMAE 2016agallafiBelum ada peringkat
- Home Mane Animal Feed Concentrate - FAO - PTG PDFDokumen12 halamanHome Mane Animal Feed Concentrate - FAO - PTG PDFjacjiBelum ada peringkat
- Ores and Metallurgy PDFDokumen38 halamanOres and Metallurgy PDFAniruddha KawadeBelum ada peringkat
- Additives Sas AsDokumen300 halamanAdditives Sas AsAnonymous OvJJxWBelum ada peringkat
- ความสามารถในการสะเทินกรดของยาลดกรด (Meq Of Hcl/G Of Antacid) 5Dokumen4 halamanความสามารถในการสะเทินกรดของยาลดกรด (Meq Of Hcl/G Of Antacid) 5Mubarak PatelBelum ada peringkat
- Painting Study NotesDokumen243 halamanPainting Study NotesAryan Kaul100% (6)
- Pilot Plant Scale Extraction of Alginate From Macrocystis PyriferaDokumen8 halamanPilot Plant Scale Extraction of Alginate From Macrocystis PyriferaMasiBelum ada peringkat
- QuantitativeDokumen29 halamanQuantitativeapi-422428700Belum ada peringkat
- Arthritis - BoronDokumen18 halamanArthritis - BoronNatalia NeliBelum ada peringkat
- MSDS Pupuk Haracoat PDFDokumen4 halamanMSDS Pupuk Haracoat PDFAnonymous UQVygYg3lfBelum ada peringkat
- BS For Chemicals To Water TreatmentDokumen18 halamanBS For Chemicals To Water TreatmentKalinda0% (1)
- Analisa AgriculturalDokumen6 halamanAnalisa AgriculturalFEBRINA SARLINDA, STBelum ada peringkat
- Nutritional Values and Pharmacological Importance of Date Fruit (Phoenix Dactylifera Linn) A ReviewDokumen4 halamanNutritional Values and Pharmacological Importance of Date Fruit (Phoenix Dactylifera Linn) A ReviewDr. Md. Shahab UddinBelum ada peringkat
- Bioinorganic ChemistryDokumen41 halamanBioinorganic ChemistryRamagouda HinglajeBelum ada peringkat
- The End of Craving: Recovering the Lost Wisdom of Eating WellDari EverandThe End of Craving: Recovering the Lost Wisdom of Eating WellPenilaian: 4.5 dari 5 bintang4.5/5 (82)
- Sully: The Untold Story Behind the Miracle on the HudsonDari EverandSully: The Untold Story Behind the Miracle on the HudsonPenilaian: 4 dari 5 bintang4/5 (103)
- Permaculture for the Rest of Us: Abundant Living on Less than an AcreDari EverandPermaculture for the Rest of Us: Abundant Living on Less than an AcrePenilaian: 4.5 dari 5 bintang4.5/5 (33)
- The Fabric of Civilization: How Textiles Made the WorldDari EverandThe Fabric of Civilization: How Textiles Made the WorldPenilaian: 4.5 dari 5 bintang4.5/5 (58)
- The Technology Trap: Capital, Labor, and Power in the Age of AutomationDari EverandThe Technology Trap: Capital, Labor, and Power in the Age of AutomationPenilaian: 4.5 dari 5 bintang4.5/5 (46)
- The Future of Geography: How the Competition in Space Will Change Our WorldDari EverandThe Future of Geography: How the Competition in Space Will Change Our WorldPenilaian: 4 dari 5 bintang4/5 (6)
- Four Battlegrounds: Power in the Age of Artificial IntelligenceDari EverandFour Battlegrounds: Power in the Age of Artificial IntelligencePenilaian: 5 dari 5 bintang5/5 (5)
- The Intel Trinity: How Robert Noyce, Gordon Moore, and Andy Grove Built the World's Most Important CompanyDari EverandThe Intel Trinity: How Robert Noyce, Gordon Moore, and Andy Grove Built the World's Most Important CompanyBelum ada peringkat
- Hero Found: The Greatest POW Escape of the Vietnam WarDari EverandHero Found: The Greatest POW Escape of the Vietnam WarPenilaian: 4 dari 5 bintang4/5 (19)
- Highest Duty: My Search for What Really MattersDari EverandHighest Duty: My Search for What Really MattersBelum ada peringkat
- System Error: Where Big Tech Went Wrong and How We Can RebootDari EverandSystem Error: Where Big Tech Went Wrong and How We Can RebootBelum ada peringkat
- A Place of My Own: The Architecture of DaydreamsDari EverandA Place of My Own: The Architecture of DaydreamsPenilaian: 4 dari 5 bintang4/5 (242)
- How to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerDari EverandHow to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerPenilaian: 4.5 dari 5 bintang4.5/5 (54)
- Fire on the Horizon: The Untold Story of the Gulf Oil DisasterDari EverandFire on the Horizon: The Untold Story of the Gulf Oil DisasterBelum ada peringkat
- Pale Blue Dot: A Vision of the Human Future in SpaceDari EverandPale Blue Dot: A Vision of the Human Future in SpacePenilaian: 4.5 dari 5 bintang4.5/5 (588)
- The Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaDari EverandThe Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaBelum ada peringkat
- ChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindDari EverandChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindBelum ada peringkat
- Dirt to Soil: One Family’s Journey into Regenerative AgricultureDari EverandDirt to Soil: One Family’s Journey into Regenerative AgriculturePenilaian: 5 dari 5 bintang5/5 (125)
- Faster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestDari EverandFaster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestPenilaian: 4 dari 5 bintang4/5 (28)
- Reality+: Virtual Worlds and the Problems of PhilosophyDari EverandReality+: Virtual Worlds and the Problems of PhilosophyPenilaian: 4 dari 5 bintang4/5 (24)
- The Assassination Complex: Inside the Government's Secret Drone Warfare ProgramDari EverandThe Assassination Complex: Inside the Government's Secret Drone Warfare ProgramPenilaian: 4 dari 5 bintang4/5 (55)
- The Manager's Path: A Guide for Tech Leaders Navigating Growth and ChangeDari EverandThe Manager's Path: A Guide for Tech Leaders Navigating Growth and ChangePenilaian: 4.5 dari 5 bintang4.5/5 (99)
- Mini Farming: Self-Sufficiency on 1/4 AcreDari EverandMini Farming: Self-Sufficiency on 1/4 AcrePenilaian: 4 dari 5 bintang4/5 (76)
- The Things We Make: The Unknown History of Invention from Cathedrals to Soda Cans (Father's Day Gift for Science and Engineering Curious Dads)Dari EverandThe Things We Make: The Unknown History of Invention from Cathedrals to Soda Cans (Father's Day Gift for Science and Engineering Curious Dads)Belum ada peringkat
- Broken Money: Why Our Financial System is Failing Us and How We Can Make it BetterDari EverandBroken Money: Why Our Financial System is Failing Us and How We Can Make it BetterPenilaian: 5 dari 5 bintang5/5 (3)
- Transformed: Moving to the Product Operating ModelDari EverandTransformed: Moving to the Product Operating ModelPenilaian: 4 dari 5 bintang4/5 (1)