Anda mungkin juga menyukai
- Distribution of DrugsDokumen37 halamanDistribution of DrugsNazmul Nabil100% (1)
- Drug DistributionDokumen49 halamanDrug DistributionRinta Moon100% (1)
- PharmacokineticsDokumen95 halamanPharmacokineticsSonalee ShahBelum ada peringkat
- PharmacokineticsDokumen112 halamanPharmacokineticsGog Rg100% (2)
- 2.one Compartment Open ModelDokumen90 halaman2.one Compartment Open ModelBLESSY SARA KURIANBelum ada peringkat
- Pharamcokinetics: Course In-Charge: Nimra Waheed Course Name: Biopharmaceutics and Pharmacokinetics Course Code: 613-TDokumen21 halamanPharamcokinetics: Course In-Charge: Nimra Waheed Course Name: Biopharmaceutics and Pharmacokinetics Course Code: 613-TNeha GulfamBelum ada peringkat
- Introduction To BiopharmaceuticsDokumen27 halamanIntroduction To BiopharmaceuticsAmina Akther Mim 1821179649Belum ada peringkat
- 1 - Pharmaceutical Care Practice - An OverviewDokumen76 halaman1 - Pharmaceutical Care Practice - An OverviewekramBelum ada peringkat
- Nonlinear Pharmacokinetics 1Dokumen34 halamanNonlinear Pharmacokinetics 1donndisaster100% (1)
- Biopharmaceutics and Clinical PharmacokineticsDokumen302 halamanBiopharmaceutics and Clinical PharmacokineticsBalisa MosisaBelum ada peringkat
- Therapeutic Drug Monitoring GuideDokumen33 halamanTherapeutic Drug Monitoring GuideDr. Raghavendra Kumar GundaBelum ada peringkat
- Excretion of Drugs: Dr. G.SAILAJA, M.Pharm - PH.D., Assoc. Professor, Dept. of PharmaceuticsDokumen43 halamanExcretion of Drugs: Dr. G.SAILAJA, M.Pharm - PH.D., Assoc. Professor, Dept. of PharmaceuticsSailaja Reddy GunnamBelum ada peringkat
- Application of PK in Clinical SitutionDokumen42 halamanApplication of PK in Clinical Situtionsafia mehmood100% (1)
- Absorption of DrugsDokumen34 halamanAbsorption of Drugsalexpharm100% (1)
- Clinical Pharmacokinetics 2013Dokumen68 halamanClinical Pharmacokinetics 2013Law YouBelum ada peringkat
- Introduction To BiopharmaceuticsDokumen106 halamanIntroduction To BiopharmaceuticsHely Patel100% (1)
- One Compartment KineticsDokumen31 halamanOne Compartment KineticsAna Francisca100% (1)
- ExcretionDokumen35 halamanExcretionHely PatelBelum ada peringkat
- Nonlinear Pharmacokinetics ExplainedDokumen39 halamanNonlinear Pharmacokinetics ExplainedYuppie RajBelum ada peringkat
- Chapter 7. Pharmacokinetics of Oral AbsorptionDokumen28 halamanChapter 7. Pharmacokinetics of Oral AbsorptionbencleeseBelum ada peringkat
- Compartment ModellingDokumen46 halamanCompartment ModellingBio DataBelum ada peringkat
- Excretion of DrugDokumen36 halamanExcretion of DrugYeni SuwitaBelum ada peringkat
- Therapeutic Drug Monitoring-FinalDokumen49 halamanTherapeutic Drug Monitoring-FinalSaiesh PhaldesaiBelum ada peringkat
- Nonlinear Pharmacokinetics 1Dokumen11 halamanNonlinear Pharmacokinetics 1Mubashar ShahidBelum ada peringkat
- Chapter 3. One-Compartment Open Model Intravenous Bolus AdministrationDokumen23 halamanChapter 3. One-Compartment Open Model Intravenous Bolus AdministrationbencleeseBelum ada peringkat
- Clinical PharmacokineticsDokumen36 halamanClinical PharmacokineticsDr.U.P.Rathnakar.MD.DIH.PGDHM80% (5)
- 07 Dosage RegimenDokumen44 halaman07 Dosage Regimenzetttttttttt100% (3)
- Volume of DistributionDokumen31 halamanVolume of DistributionMinal Nadeem100% (1)
- Bioavailability and BioequivalenceDokumen82 halamanBioavailability and Bioequivalenceكسلان اكتب اسمي100% (1)
- MDR Repetitive Intravenous InjectionsDokumen17 halamanMDR Repetitive Intravenous InjectionsMuhammad MursaleenBelum ada peringkat
- IV Infusion Methods for Calculating Patient Elimination Half-LifeDokumen20 halamanIV Infusion Methods for Calculating Patient Elimination Half-Lifeكسلان اكتب اسمي100% (1)
- Unit IV. Solid Modified Release Dosage FormsDokumen24 halamanUnit IV. Solid Modified Release Dosage FormsMary-Ann Valencia SapnuBelum ada peringkat
- Clinical PharmacokineticsDokumen36 halamanClinical PharmacokineticsWalaa YousefBelum ada peringkat
- Medication Order ReviewDokumen34 halamanMedication Order ReviewVidho RiveraBelum ada peringkat
- Hepatic ClearanceDokumen3 halamanHepatic ClearanceMiami Arif100% (1)
- Non Linear PharmacokineticsDokumen94 halamanNon Linear PharmacokineticsJaspreet Guraya100% (1)
- 1 Introduction - BiopharmaceuticsDokumen32 halaman1 Introduction - BiopharmaceuticsLouie Fernand LegaspiBelum ada peringkat
- Parenteral Preparation PDFDokumen17 halamanParenteral Preparation PDFHely Patel100% (1)
- Pharmacokinetics: Dr. Jahid MBBS, M.phil (Pharmacology) Head of Pharmacology (MD-AUCMS)Dokumen52 halamanPharmacokinetics: Dr. Jahid MBBS, M.phil (Pharmacology) Head of Pharmacology (MD-AUCMS)vivianBelum ada peringkat
- Absorption of DrugsDokumen41 halamanAbsorption of DrugsSibtain100% (2)
- CalculationDokumen24 halamanCalculationhablet1100% (1)
- BiopharmaceuticsDokumen52 halamanBiopharmaceuticsDharma ShantiniBelum ada peringkat
- Pharmacokinetics: Saminathan Kayarohanam M.Pharm, M.B.A, PHDDokumen26 halamanPharmacokinetics: Saminathan Kayarohanam M.Pharm, M.B.A, PHDSaminathan KayarohanamBelum ada peringkat
- Introduction to Biopharmaceutics & PharmacokineticsDokumen34 halamanIntroduction to Biopharmaceutics & PharmacokineticsMd. Yasir Galib 1721609649100% (1)
- Compartment ModelsDokumen67 halamanCompartment ModelsSunil100% (3)
- Pharmacotherapeutics UNIT1Dokumen44 halamanPharmacotherapeutics UNIT1Bharti ChauhanBelum ada peringkat
- Clinical PharmacokineticsDokumen31 halamanClinical PharmacokineticsArdiyanti Puspitasari100% (1)
- Adverse Reactions SlideshowDokumen40 halamanAdverse Reactions SlideshowGary MaoBelum ada peringkat
- Drug ExcretionDokumen20 halamanDrug ExcretionRajalingam BalaBelum ada peringkat
- Drug Extraction RatioDokumen8 halamanDrug Extraction RatioHeru Fajar SyaputraBelum ada peringkat
- Emulsion and ApplicationDokumen26 halamanEmulsion and ApplicationSamay Sharma100% (1)
- Prelim (Drug Delivery System)Dokumen133 halamanPrelim (Drug Delivery System)Vanessa DL100% (1)
- Therapeutic Drug Monitoring 7-1-20Dokumen27 halamanTherapeutic Drug Monitoring 7-1-20mofadhilBelum ada peringkat
- Lecture PharmacokineticsDokumen55 halamanLecture Pharmacokineticssatya narayan100% (2)
- Nonlinear Pharmacokinetics: Guided By: Dhaivat C. ParikhDokumen46 halamanNonlinear Pharmacokinetics: Guided By: Dhaivat C. ParikhTushar Bambharoliya100% (5)
- Agents Used in Blood DisordersDokumen54 halamanAgents Used in Blood DisordersJing Lomboy AcostaBelum ada peringkat
- General Pharmacology (Distribution)Dokumen47 halamanGeneral Pharmacology (Distribution)rexef82757Belum ada peringkat
- Farkin 1Dokumen9 halamanFarkin 1Yunita Carolline PanggabeanBelum ada peringkat
- Distribution of Drugs: Distribution Is Defined As The Reversible Transfer of Drugs Between Body Fluid CompartmentsDokumen9 halamanDistribution of Drugs: Distribution Is Defined As The Reversible Transfer of Drugs Between Body Fluid Compartmentsdavid5king-3119Belum ada peringkat
- PharmacokineticsDokumen95 halamanPharmacokineticsshripathyd1100% (1)
- Drugs in ReptilesDokumen71 halamanDrugs in ReptilesSunil100% (1)
- Household Hazards To PetsDokumen16 halamanHousehold Hazards To PetsSunilBelum ada peringkat
- ENDOCRINE PHARMACOLOGY: THYROID AND PANCREATIC HORMONESDokumen16 halamanENDOCRINE PHARMACOLOGY: THYROID AND PANCREATIC HORMONESSunilBelum ada peringkat
- Mycotoxins and Bacterial Toxins: Types, Characteristics and EffectsDokumen103 halamanMycotoxins and Bacterial Toxins: Types, Characteristics and EffectsSunil100% (1)
- Veterinary Pharmacology and Toxicology MCQsDokumen10 halamanVeterinary Pharmacology and Toxicology MCQsSunilBelum ada peringkat
- Classification and Dosage of Antimicrobial Agents in Veterinary MedicineDokumen24 halamanClassification and Dosage of Antimicrobial Agents in Veterinary MedicineSunil0% (1)
- Classification of AmphibiansDokumen22 halamanClassification of AmphibiansSunilBelum ada peringkat
- Carbon Monoxide PoisoningDokumen22 halamanCarbon Monoxide PoisoningSunilBelum ada peringkat
- FLUORINE TOXICITY AND FLUOROSISDokumen55 halamanFLUORINE TOXICITY AND FLUOROSISSunilBelum ada peringkat
- Drugs Acting On Respiratory System of AnimalsDokumen8 halamanDrugs Acting On Respiratory System of AnimalsSunil100% (4)
- Toxicological Investigation and Its Significance in Animal Health DiagnosisDokumen8 halamanToxicological Investigation and Its Significance in Animal Health DiagnosisSunilBelum ada peringkat
- Drugs in Behavioural Disorders of PetsDokumen5 halamanDrugs in Behavioural Disorders of PetsSunilBelum ada peringkat
- Antiseptics and Disinfectants For Veterinary ClinicsDokumen3 halamanAntiseptics and Disinfectants For Veterinary ClinicsSunil100% (5)
- VCI MSVE 2008 RegulationsDokumen136 halamanVCI MSVE 2008 RegulationsSunil50% (2)
- Drugs Acting On Haematopoietic System of AnimalsDokumen28 halamanDrugs Acting On Haematopoietic System of AnimalsSunil100% (1)
- Nsaids and Other Antinflammatory Agents in Veterinary PracticeDokumen44 halamanNsaids and Other Antinflammatory Agents in Veterinary PracticeSunil100% (2)
- Drugs Acting On Genitourinary System of AnimalsDokumen44 halamanDrugs Acting On Genitourinary System of AnimalsSunilBelum ada peringkat
- Fluorosis Phosphorous ToxicityDokumen55 halamanFluorosis Phosphorous ToxicitySunilBelum ada peringkat
- Drugs Acting On Digestive System of AnimalsDokumen11 halamanDrugs Acting On Digestive System of AnimalsSunil100% (3)
- GENERAL LINE of Treatment UREA AMMONIA SALT - poISONINGDokumen49 halamanGENERAL LINE of Treatment UREA AMMONIA SALT - poISONINGSunil0% (1)
- Mercury Lead Arsenic Cadmium ToxicityDokumen171 halamanMercury Lead Arsenic Cadmium ToxicitySunilBelum ada peringkat
- Behavioural Modifying Drugs in PetsDokumen33 halamanBehavioural Modifying Drugs in PetsSunilBelum ada peringkat
- LATHYRISM AND PHOTOSENSiTIZATIONDokumen33 halamanLATHYRISM AND PHOTOSENSiTIZATIONSunilBelum ada peringkat
- Toxicokinetics DynamicsDokumen76 halamanToxicokinetics DynamicsSunil100% (1)
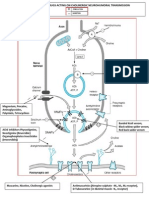
- Site of Action of Drugs Acting On Cholinergic Neurohumoral Transmission PDFDokumen1 halamanSite of Action of Drugs Acting On Cholinergic Neurohumoral Transmission PDFSunilBelum ada peringkat
- Environmental PollutantsDokumen32 halamanEnvironmental PollutantsSunilBelum ada peringkat
- ZOOTOXINSDokumen72 halamanZOOTOXINSSunil83% (6)
- Mycotoxins and Bacterial Toxins: Types, Characteristics and EffectsDokumen103 halamanMycotoxins and Bacterial Toxins: Types, Characteristics and EffectsSunil100% (1)
- Site of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFDokumen1 halamanSite of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFSunilBelum ada peringkat
- Epilepsy and Anticonvulsant DrugsDokumen28 halamanEpilepsy and Anticonvulsant DrugsSunilBelum ada peringkat
- COE WorksheetDokumen16 halamanCOE WorksheetiloveraynaBelum ada peringkat
- Nutrition FactsDokumen232 halamanNutrition FactsAriana Sălăjan100% (2)
- How To Write A Systematic ReviewDokumen8 halamanHow To Write A Systematic ReviewNader Alaizari100% (1)
- Varicella in Pregnancy GuidelineDokumen5 halamanVaricella in Pregnancy GuidelineYwagar YwagarBelum ada peringkat
- The Tuskegee Syphilis StudyDokumen11 halamanThe Tuskegee Syphilis Studyapi-285171544Belum ada peringkat
- Do Clear Cell Ovarian Carcinomas Have Poorer Prognosis Compared To Other Epithelial Cell Types? A Study of 1411 Clear Cell Ovarian CancersDokumen7 halamanDo Clear Cell Ovarian Carcinomas Have Poorer Prognosis Compared To Other Epithelial Cell Types? A Study of 1411 Clear Cell Ovarian CancersNi Wayan Ana PsBelum ada peringkat
- FurosemideDokumen4 halamanFurosemideapi-3797941100% (1)
- Perinatal DepressionDokumen200 halamanPerinatal DepressionPretty PuspitasariBelum ada peringkat
- Potential Long-Term Side Effects of Exposure To ChemoDokumen21 halamanPotential Long-Term Side Effects of Exposure To ChemoMichael BubleBelum ada peringkat
- Emergencies in GITDokumen7 halamanEmergencies in GITsssajiBelum ada peringkat
- The Management of Near DrowningDokumen8 halamanThe Management of Near DrowningneddiantiseptikaBelum ada peringkat
- Premature Rupture of MembranesDokumen5 halamanPremature Rupture of MembranesJoselyn San MiguelBelum ada peringkat
- Código Descripción Costo Unita. Existencia Unidades Costo Existencia Precio 1 %utilDokumen24 halamanCódigo Descripción Costo Unita. Existencia Unidades Costo Existencia Precio 1 %utilNiky Dos SantosBelum ada peringkat
- Gout and Hyperuricemia: PathophysiologyDokumen1 halamanGout and Hyperuricemia: Pathophysiologyسمرة طايبBelum ada peringkat
- Catalogue Wastewater Treatment SolutionsDokumen49 halamanCatalogue Wastewater Treatment SolutionsmentolBelum ada peringkat
- Sfbtnarr 2Dokumen19 halamanSfbtnarr 2api-267074391Belum ada peringkat
- Nutrition Questionnaire-Focused PregnancyDokumen2 halamanNutrition Questionnaire-Focused PregnancyJing Cruz100% (1)
- La Indications, Contraindications&Armamentarium - ppt2Dokumen25 halamanLa Indications, Contraindications&Armamentarium - ppt2Nagendra Srinivas ChunduriBelum ada peringkat
- Red EyesDokumen3 halamanRed EyesirijoaBelum ada peringkat
- Dim Mak and Acupuncture: Understanding the Medical Reasons Behind this Martial ArtDokumen6 halamanDim Mak and Acupuncture: Understanding the Medical Reasons Behind this Martial ArtIoan NeacsuBelum ada peringkat
- Data Dictionary: NO Label Operational Scale of Measurem ENT 1Dokumen6 halamanData Dictionary: NO Label Operational Scale of Measurem ENT 1nik nur nisa azlinBelum ada peringkat
- Konversi Kode Prosedure INACBGs Ke Kode ICD 9-CMDokumen349 halamanKonversi Kode Prosedure INACBGs Ke Kode ICD 9-CMTri Muhammad HaniBelum ada peringkat
- AHA ACLS Provider Manual $45Dokumen2 halamanAHA ACLS Provider Manual $45Jigar GandhiBelum ada peringkat
- Javma-Javma 21 04 0213Dokumen5 halamanJavma-Javma 21 04 0213Black manBelum ada peringkat
- Jack Poylangada - Copy of Stamper Self-Care JournalDokumen8 halamanJack Poylangada - Copy of Stamper Self-Care Journalapi-503633296100% (3)
- 1000 Days Ni Baby PDFDokumen31 halaman1000 Days Ni Baby PDFppantollanaBelum ada peringkat
- Nursing ProcessDokumen118 halamanNursing Processɹǝʍdןnos100% (30)
- LTCW Breast Write UpDokumen21 halamanLTCW Breast Write Upapi-632682404Belum ada peringkat
- Humalog - Uses, Dosage & Side Effects - DrugsDokumen4 halamanHumalog - Uses, Dosage & Side Effects - Drugsremyde07100% (1)
- Measuring Lung Capacity PDFDokumen5 halamanMeasuring Lung Capacity PDFSip BioBelum ada peringkat