Anda mungkin juga menyukai
- Gastroretentive Drug Delivery SystemDokumen7 halamanGastroretentive Drug Delivery SystemAdvanced Research PublicationsBelum ada peringkat
- Formulation and Evaluation of Anti Emetic Patch Comprising On Dan Set Ron Hydro ChlorideDokumen107 halamanFormulation and Evaluation of Anti Emetic Patch Comprising On Dan Set Ron Hydro ChlorideuduriskBelum ada peringkat
- HPLC Pharma 25-1-09-Numbered-All PrintDokumen19 halamanHPLC Pharma 25-1-09-Numbered-All PrintshulalevinBelum ada peringkat
- FDA - ICH M7 (R1) - Control of Mutagenic Impurities in Pharmaceuticals 03.2018Dokumen131 halamanFDA - ICH M7 (R1) - Control of Mutagenic Impurities in Pharmaceuticals 03.2018Catrinescu OanaBelum ada peringkat
- Transdermal Drug Delivery: BackingDokumen32 halamanTransdermal Drug Delivery: BackingmurtazaBelum ada peringkat
- Insitu Gels PaperDokumen7 halamanInsitu Gels PaperShreya100% (1)
- Osmotic Drug Delivery SystemDokumen10 halamanOsmotic Drug Delivery SystemkishoreBelum ada peringkat
- Penetration EnhancersDokumen17 halamanPenetration EnhancersPharmaosmosis NiperBelum ada peringkat
- BASIC PHARMACOKINETICS - CHAPTER 5: IV InfusionDokumen42 halamanBASIC PHARMACOKINETICS - CHAPTER 5: IV InfusionDrHeba100% (6)
- Homology Modelling Notes PDFDokumen30 halamanHomology Modelling Notes PDFkamleshBelum ada peringkat
- Transdermal Drug Delivery System An OverviewDokumen10 halamanTransdermal Drug Delivery System An OverviewJoko RinantoBelum ada peringkat
- Design of Dosage FormsDokumen17 halamanDesign of Dosage FormsMuhammad HilmiBelum ada peringkat
- Transdermal Drug Delivery SystemDokumen47 halamanTransdermal Drug Delivery SystemMridul AroraBelum ada peringkat
- EMLA Product MonographDokumen46 halamanEMLA Product Monographketan79797Belum ada peringkat
- Pharmaceutics Chapter 7 Novel Drug Delivery System NotesDokumen10 halamanPharmaceutics Chapter 7 Novel Drug Delivery System NotesBhuvnesh ChandraBelum ada peringkat
- Use and Limitations of in Vitro Dissolution Testing: Topic Introduction and OverviewDokumen114 halamanUse and Limitations of in Vitro Dissolution Testing: Topic Introduction and OverviewMuthu Venkatesh100% (1)
- HPLC ColumnDokumen9 halamanHPLC Columnbayu permanaBelum ada peringkat
- Topical GelDokumen4 halamanTopical GelBINDUBelum ada peringkat
- Fundamental Lc-Ms IntroductionDokumen25 halamanFundamental Lc-Ms IntroductionAndreaAbdelLatifBelum ada peringkat
- ANTI INFLAMMATORY Screening MethodsDokumen7 halamanANTI INFLAMMATORY Screening MethodsBrajesh Thankamony67% (3)
- Liquid Chromatography Mass Spectrometry inDokumen50 halamanLiquid Chromatography Mass Spectrometry inGiray ErsözoğluBelum ada peringkat
- Osmotic Drug Delivery Systems 3Dokumen48 halamanOsmotic Drug Delivery Systems 3anupnakat50% (2)
- Transdermal Nano BookDokumen44 halamanTransdermal Nano BookMuhammad Azam TahirBelum ada peringkat
- Pharmaceutical Analysis Syllabus in NIPERDokumen12 halamanPharmaceutical Analysis Syllabus in NIPERHARI HARA RAO GUJJARBelum ada peringkat
- XRD ReportDokumen13 halamanXRD ReportMukulBelum ada peringkat
- GRDDSDokumen31 halamanGRDDSMuhammad Azam TahirBelum ada peringkat
- Applications of Eutectic Mixtures in Pharmaceutical IndustryDokumen3 halamanApplications of Eutectic Mixtures in Pharmaceutical IndustryKevin CruzBelum ada peringkat
- HPTLC SeminarDokumen23 halamanHPTLC SeminarAzim Arshi100% (9)
- Drug Metabolism - Chapter 8Dokumen44 halamanDrug Metabolism - Chapter 8Shaun李好Belum ada peringkat
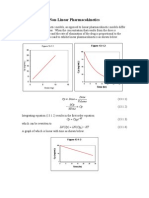
- BASIC PHARMACOKINETICS - CHAPTER 13: Non-Linear KineticsDokumen22 halamanBASIC PHARMACOKINETICS - CHAPTER 13: Non-Linear KineticsDrHeba100% (7)
- Liposomes, Niosomes and NanotechnologyDokumen60 halamanLiposomes, Niosomes and Nanotechnologyrksinghjjn100% (1)
- Oral Mucosal Drug Delivery and TherapyDokumen289 halamanOral Mucosal Drug Delivery and TherapyАлександр ВолошанBelum ada peringkat
- Application of HPLCDokumen11 halamanApplication of HPLCIts KazmiBelum ada peringkat
- BASIC PHARMACOKINETICS - CHAPTER 15: Exams IIDokumen5 halamanBASIC PHARMACOKINETICS - CHAPTER 15: Exams IIDrHeba100% (1)
- Sustained Release Drug Delivery System - A ReviewDokumen14 halamanSustained Release Drug Delivery System - A ReviewPranav PatelBelum ada peringkat
- Floating FilmDokumen13 halamanFloating Filmchittam suvarnaBelum ada peringkat
- Dermato ToxicologyDokumen1.121 halamanDermato ToxicologyBella LunaBelum ada peringkat
- Polymeric Drugs and Drug Delivery SystemDokumen313 halamanPolymeric Drugs and Drug Delivery SystemEllahnae VelasquezBelum ada peringkat
- Advances in Chemical Engineering, Volume 29 - Molecular and Cellular Foundations of BiomaterialsDokumen211 halamanAdvances in Chemical Engineering, Volume 29 - Molecular and Cellular Foundations of BiomaterialsRedV1rusBelum ada peringkat
- Ravi Pratap Pulla: Asso - Professor, SSJ College of Pharmacy, V.N.Pally, Gandipet, Hyderabad-75Dokumen304 halamanRavi Pratap Pulla: Asso - Professor, SSJ College of Pharmacy, V.N.Pally, Gandipet, Hyderabad-75Ravi Pratap Pulla100% (2)
- UV SpectrosDokumen4 halamanUV SpectrosCarlton GrantBelum ada peringkat
- Transdermal Drug DeliveryDokumen73 halamanTransdermal Drug DeliveryHairuddin To100% (2)
- Biopharmaceutics and Pharmacokinetics P.L.Madan PDFDokumen450 halamanBiopharmaceutics and Pharmacokinetics P.L.Madan PDFSyeda Urooj Fatima40% (5)
- Mass Spectrometry-Based Proteomics PDFDokumen10 halamanMass Spectrometry-Based Proteomics PDFDomenico StaianoBelum ada peringkat
- BASIC PHARMACOKINETICS - CHAPTER 7: Oral DosingDokumen80 halamanBASIC PHARMACOKINETICS - CHAPTER 7: Oral DosingDrHeba50% (2)
- Supercritical Fluid Chromatography (SFC)Dokumen32 halamanSupercritical Fluid Chromatography (SFC)masruri123Belum ada peringkat
- Lecture5 Pharmaceutics (Buffer Partition)Dokumen6 halamanLecture5 Pharmaceutics (Buffer Partition)haroon41Belum ada peringkat
- Formulation and Evaluation ofDokumen289 halamanFormulation and Evaluation ofThuhieu HuaBelum ada peringkat
- Topical Drug Delivery SystemsDokumen33 halamanTopical Drug Delivery SystemsIulia July100% (1)
- Transdermal Drug Delivery Systems (An Overview) PDFDokumen9 halamanTransdermal Drug Delivery Systems (An Overview) PDFCheng BauzonBelum ada peringkat
- Polymers in Pharmaceutical ProductsDokumen14 halamanPolymers in Pharmaceutical Productsstudent910112Belum ada peringkat
- Absorbance and Fluorescence Spectroscopies of Green Fluorescent ProteinDokumen24 halamanAbsorbance and Fluorescence Spectroscopies of Green Fluorescent ProteinMadel Tutor ChaturvediBelum ada peringkat
- Guideline Quality Transdermal Patches enDokumen27 halamanGuideline Quality Transdermal Patches endinaBelum ada peringkat
- Enzyme AssayDokumen23 halamanEnzyme AssayTanvir JawadBelum ada peringkat
- A Prolonged Release Parenteral Drug Delivery SystemDokumen11 halamanA Prolonged Release Parenteral Drug Delivery SystemronnymcmBelum ada peringkat
- Form 28 After A4vDokumen3 halamanForm 28 After A4vWynette Winny Williams100% (7)
- Sholarship Letter - Doc FormatDokumen6 halamanSholarship Letter - Doc FormatAnirudh Jain ChampawatBelum ada peringkat
- Industry Visit Reqeust Letter FormatDokumen1 halamanIndustry Visit Reqeust Letter FormatAnirudh Jain ChampawatBelum ada peringkat
- Exp Letter FormatDokumen1 halamanExp Letter FormatAnirudh Jain ChampawatBelum ada peringkat
- Spectrum PHARMDokumen17 halamanSpectrum PHARMAnirudh Jain ChampawatBelum ada peringkat
- Gujarat Pharma08Dokumen29 halamanGujarat Pharma08Anirudh Jain ChampawatBelum ada peringkat
- M.Pharm Thesis Aim of The PresentDokumen1 halamanM.Pharm Thesis Aim of The PresentAnirudh Jain ChampawatBelum ada peringkat
- Paarenteral ProductionDokumen32 halamanPaarenteral ProductionHanuma KanthetiBelum ada peringkat
- Drugs & Dosage FormsDokumen25 halamanDrugs & Dosage FormsSenoadji Pratama100% (1)
- Nursing Skills Checklist For 2103 3104 12-10-12Dokumen6 halamanNursing Skills Checklist For 2103 3104 12-10-12api-279087520Belum ada peringkat
- Fundamentals of NddsDokumen26 halamanFundamentals of NddsPavan NagdevBelum ada peringkat
- Oral DDF 3 2 Gupta PDFDokumen13 halamanOral DDF 3 2 Gupta PDFZaki AhmadBelum ada peringkat
- B.Sc. (N) SyllabusDokumen169 halamanB.Sc. (N) SyllabusMUTHUKUMARANBelum ada peringkat
- Excipient-Free Pulmonary Insulin Dry PowderDokumen9 halamanExcipient-Free Pulmonary Insulin Dry PowderYaninaDeLafuenteBelum ada peringkat
- AntibioticsDokumen16 halamanAntibioticsaattaa100% (2)
- Fundamentals of NursingDokumen4 halamanFundamentals of NursingLala DunqueBelum ada peringkat
- Etextbook PDF For Juliens Primer of Drug Action A Comprehensive Guide To The Actions Uses and Side Effects of Psychoactive Drugs 14th EditioDokumen62 halamanEtextbook PDF For Juliens Primer of Drug Action A Comprehensive Guide To The Actions Uses and Side Effects of Psychoactive Drugs 14th Editiobeverly.bustos487100% (42)
- Simulate SubQ - The Methods and The MediaDokumen17 halamanSimulate SubQ - The Methods and The MediaLaudaaBelum ada peringkat
- Chapter 12: Medication Dosage Forms and Routes of AdministrationDokumen29 halamanChapter 12: Medication Dosage Forms and Routes of AdministrationJana EncaboBelum ada peringkat
- Elemental Impurities PDFDokumen85 halamanElemental Impurities PDFsuey0209Belum ada peringkat
- Introduction To Nursing PharmacologyDokumen97 halamanIntroduction To Nursing PharmacologyLiel TorresBelum ada peringkat
- NCM 106 - Pharmacology Midterms 1M-4M Edited PDFDokumen43 halamanNCM 106 - Pharmacology Midterms 1M-4M Edited PDFKisha Bethel100% (2)
- EndotoxinLimits SeanJH PR17 012Dokumen7 halamanEndotoxinLimits SeanJH PR17 012sppBelum ada peringkat
- Phases of Drug DevelopmentDokumen14 halamanPhases of Drug DevelopmentMabesBelum ada peringkat
- Clinical Pharmacokinetics Theophyllin: Kelas A/Kelompok 12Dokumen33 halamanClinical Pharmacokinetics Theophyllin: Kelas A/Kelompok 12DelisaPutriChaniagoBelum ada peringkat
- Pain Relief in Palliative Care, A Focus On Interventional Pain Management PDFDokumen11 halamanPain Relief in Palliative Care, A Focus On Interventional Pain Management PDFShinichi Ferry RoferdiBelum ada peringkat
- Notes On Pharmacy Services On NC2Dokumen57 halamanNotes On Pharmacy Services On NC2Lesly LogartaBelum ada peringkat
- Quiz Lecture 2Dokumen4 halamanQuiz Lecture 2Hoang Uyen Vy NguyenBelum ada peringkat
- 4 - Drug Product Design ParametersDokumen7 halaman4 - Drug Product Design ParametersVinz AlvarezBelum ada peringkat
- Unit 3, Novel Drug Delivery Systems, B Pharmacy 7th Sem, Carewell PharmaDokumen33 halamanUnit 3, Novel Drug Delivery Systems, B Pharmacy 7th Sem, Carewell Pharmaayush pathak100% (1)
- Buscopan Ta BinjDokumen6 halamanBuscopan Ta BinjNashaat H. AlshawabkehBelum ada peringkat
- Patent 1970 PDFDokumen152 halamanPatent 1970 PDFMukesh ShuklaBelum ada peringkat
- Preparation and Evaluation of Sustained Release Microbeads of Norfloxacin Using Sodium AlginateDokumen5 halamanPreparation and Evaluation of Sustained Release Microbeads of Norfloxacin Using Sodium AlginateRajesh KumarBelum ada peringkat
- Nursing Skills ChecklistDokumen11 halamanNursing Skills Checklistapi-346051113Belum ada peringkat
- Prescribing Information and Patient Medication Information: BuscopanDokumen27 halamanPrescribing Information and Patient Medication Information: BuscopanMey KhBelum ada peringkat
- Ratio and Proportion Dosage Calculations 2nd Edition Giangrasso Solutions ManualDokumen36 halamanRatio and Proportion Dosage Calculations 2nd Edition Giangrasso Solutions Manualcostardrivel.4vwhu100% (23)
- Drug AdministrationDokumen63 halamanDrug Administrationabdisalaan hassanBelum ada peringkat