Anda mungkin juga menyukai
- Acidosis MetabólicaDokumen23 halamanAcidosis MetabólicaDaniel Almazán MonroyBelum ada peringkat
- Catalogo Omnilife Peru 2021Dokumen34 halamanCatalogo Omnilife Peru 2021Ricardo Manosalva0% (1)
- Metabolismo Del HierroDokumen17 halamanMetabolismo Del HierroYiliana RodriguezBelum ada peringkat
- Programacion Metabolica TempranaDokumen12 halamanProgramacion Metabolica TempranaLuis LeonardoBelum ada peringkat
- Indicadores BioquímicosDokumen151 halamanIndicadores BioquímicosRodrigo Romero PichardoBelum ada peringkat
- Los Paraísos PerdidasDokumen29 halamanLos Paraísos PerdidasMariana MartínezBelum ada peringkat
- Errores innatos en el metabolismo: Un abordaje integral del diagnóstico al tratamientoDari EverandErrores innatos en el metabolismo: Un abordaje integral del diagnóstico al tratamientoBelum ada peringkat
- Entendiendo las metabolopatías: Una guía sencilla con ejemplosDari EverandEntendiendo las metabolopatías: Una guía sencilla con ejemplosBelum ada peringkat
- Vitaminas y MineralesDokumen34 halamanVitaminas y MineralesJ Carlos Meza CalderonBelum ada peringkat
- 08 HomocistinuriaDokumen45 halaman08 HomocistinuriaMari CarmenBelum ada peringkat
- Unidad 3. Actividad 2. Práctica de Laboratorio Configurar El Switch Cisco 2960Dokumen14 halamanUnidad 3. Actividad 2. Práctica de Laboratorio Configurar El Switch Cisco 2960J Manuel BuenoBelum ada peringkat
- La MalnutricionDokumen39 halamanLa MalnutricionEmily Vergara PliegoBelum ada peringkat
- Manual para entender y tratar el asma: Consejos para mejorar la calidad de vidaDari EverandManual para entender y tratar el asma: Consejos para mejorar la calidad de vidaBelum ada peringkat
- Cancer InfantilDokumen16 halamanCancer InfantilGUTIERREZ RAMIREZ WENDY LAURA 201701230 ESTUDIANTEBelum ada peringkat
- Enfermedad CeliacaDokumen21 halamanEnfermedad CeliacaMiriam LozoyaBelum ada peringkat
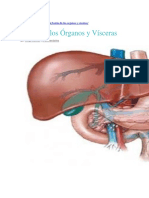
- Teoría Órganos y VíscerasDokumen6 halamanTeoría Órganos y VíscerasArq concBelum ada peringkat
- Productos Finales de Glicación Avanzada y El Riñón FinalDokumen19 halamanProductos Finales de Glicación Avanzada y El Riñón FinalVladimir HernandezBelum ada peringkat
- Encefalopatia HiperbilirrubinemicaDokumen10 halamanEncefalopatia HiperbilirrubinemicaIvantrosky MachukBelum ada peringkat
- Homocistinuria - EOOJADokumen28 halamanHomocistinuria - EOOJAnormanbaide3Belum ada peringkat
- Dieta en La Esclerosis MúltipleDokumen6 halamanDieta en La Esclerosis Múltiplejrpsaavedra4599100% (1)
- SPW Prader WillisDokumen5 halamanSPW Prader WillisMax OregliaBelum ada peringkat
- Oscilador GunnDokumen4 halamanOscilador GunnoverandresBelum ada peringkat
- Errores Innatos Del Metabolismo de Los Carbohidratos. Fructosa I, 6 DifosfatazaDokumen47 halamanErrores Innatos Del Metabolismo de Los Carbohidratos. Fructosa I, 6 DifosfatazaKaren Oscco0% (1)
- Errores Innatos Del MetabolismoDokumen18 halamanErrores Innatos Del MetabolismoSilvia Jiménez PérezBelum ada peringkat
- GRR GalactosemiaDokumen13 halamanGRR GalactosemiaAnonymous 3uLfGPBelum ada peringkat
- Metabolismo de Carbohidratos 2Dokumen8 halamanMetabolismo de Carbohidratos 2api-3696656100% (4)
- HipertrigliceridemiaDokumen35 halamanHipertrigliceridemiawww.melyBelum ada peringkat
- Encefalopatía DiabéticaDokumen2 halamanEncefalopatía DiabéticaSabrina NicoleBelum ada peringkat
- HIPERTIROIDISMO E HIPOTIROIDISMO FDokumen63 halamanHIPERTIROIDISMO E HIPOTIROIDISMO FJesus Bernabe BonillaBelum ada peringkat
- PPS Cardiologia 3 - 05 - 2016Dokumen5 halamanPPS Cardiologia 3 - 05 - 2016BI10632Belum ada peringkat
- Crecimiento y Desarrollo Del Niño Normal Grupo 5Dokumen83 halamanCrecimiento y Desarrollo Del Niño Normal Grupo 5Helen Domenica Apolo SigchoBelum ada peringkat
- Estres MetabólicoDokumen58 halamanEstres MetabólicoSandra RoriguezBelum ada peringkat
- Enfermedad de WolmanDokumen4 halamanEnfermedad de WolmanNerylyn GonzálezBelum ada peringkat
- Enfermedad de KrabbeDokumen15 halamanEnfermedad de KrabbeRicardo MembreñoBelum ada peringkat
- Enfermedad de WolmanDokumen3 halamanEnfermedad de WolmanBrayan BennettBelum ada peringkat
- Marco TeoricoDokumen9 halamanMarco TeoricoThalia HuapayaBelum ada peringkat
- Hiperlipidemia Familiar CombinadaDokumen2 halamanHiperlipidemia Familiar CombinadaDavid ArteagaBelum ada peringkat
- Encefalopatia HepaticaDokumen44 halamanEncefalopatia HepaticaWilder SantiagoBelum ada peringkat
- Errores Innatos Del MetabolismoDokumen56 halamanErrores Innatos Del MetabolismoAlgutcaBelum ada peringkat
- Dietoterapia en Insuficiencia RenalDokumen7 halamanDietoterapia en Insuficiencia RenalKatty ConsuegraBelum ada peringkat
- Grupo 8 - ZINC (Artículo de RevisiónDokumen10 halamanGrupo 8 - ZINC (Artículo de RevisiónCelier Vargas NogalesBelum ada peringkat
- Regulación de La GlucemiaDokumen10 halamanRegulación de La GlucemiaPao CabreraBelum ada peringkat
- Mecanismo de Transporte de GlucosaDokumen4 halamanMecanismo de Transporte de GlucosaBrenda TorresBelum ada peringkat
- GLUCAGONDokumen23 halamanGLUCAGONVictor Hugo Urrutia Baca100% (1)
- Deficit de Piruvato CarboxilsaDokumen3 halamanDeficit de Piruvato CarboxilsaAndrea Stefanía CuevaBelum ada peringkat
- AnorexiaDokumen8 halamanAnorexiaRubenBelum ada peringkat
- Problemas Causados Por Una Mala AlimentaciónDokumen29 halamanProblemas Causados Por Una Mala AlimentaciónElizabeth Lopez100% (4)
- Alteraciones NutricionalesDokumen5 halamanAlteraciones NutricionaleselianaBelum ada peringkat
- Síndrome de Intestino Corto. Alexis Apunte R.Dokumen7 halamanSíndrome de Intestino Corto. Alexis Apunte R.Alexis XavierBelum ada peringkat
- Presentacion Traida Del Atleta Enrique JimenezDokumen24 halamanPresentacion Traida Del Atleta Enrique JimenezEnriqueJimenezBelum ada peringkat
- Discurso Expositivo ParkinsonDokumen3 halamanDiscurso Expositivo ParkinsonMaferg ProvidaBelum ada peringkat
- Herencia Autosómica DominanteDokumen18 halamanHerencia Autosómica DominanteCC100% (1)
- GalactosemiaDokumen10 halamanGalactosemiaCsc Ana PaolaBelum ada peringkat
- Obesidad y Actividad Fisica en Los Estudiantes de La UPNFM PDFDokumen55 halamanObesidad y Actividad Fisica en Los Estudiantes de La UPNFM PDFpsiclopsBelum ada peringkat
- Enfermedad de PompeDokumen24 halamanEnfermedad de PompeValeria Castro100% (1)
- Programa de Bioclinica IIDokumen14 halamanPrograma de Bioclinica IIEsteban VegaBelum ada peringkat
- Nefropatías GlomerularesDokumen34 halamanNefropatías GlomerularesAlejandro HernándezBelum ada peringkat
- Alteraciones Del Metabolismo de La GalactosaDokumen13 halamanAlteraciones Del Metabolismo de La GalactosaRocio AlmanzaBelum ada peringkat
- Estrategia Mundial Sobre Régimen Alimentario, Actividad Física y SaludDokumen16 halamanEstrategia Mundial Sobre Régimen Alimentario, Actividad Física y SaludMariam AlemánBelum ada peringkat
- HematopoyesisDokumen5 halamanHematopoyesisPolidoro Montesinos MoguelBelum ada peringkat
- Abordaje Nutricional en El Tratamiento de La DisfagiaDokumen7 halamanAbordaje Nutricional en El Tratamiento de La DisfagiaRosy CjBelum ada peringkat
- Fisiopatología de La FenilcetonuriaDokumen1 halamanFisiopatología de La FenilcetonuriaCristina Andrea Palma CórdovaBelum ada peringkat
- Hipertensión Pulmonar Arterial: (Ascitis) En Pollos De EngordaDari EverandHipertensión Pulmonar Arterial: (Ascitis) En Pollos De EngordaBelum ada peringkat
- Apuntes sobre las necesidades nutricionales en las distantes edadesDari EverandApuntes sobre las necesidades nutricionales en las distantes edadesBelum ada peringkat
- Taller Biquimica Tercer CorteDokumen7 halamanTaller Biquimica Tercer CorteDaniela NarvaezBelum ada peringkat
- 3 Profilaxis Gastritis AgudaDokumen3 halaman3 Profilaxis Gastritis AgudaMariana Martínez0% (1)
- Caso de Osteodistrofia RenalDokumen10 halamanCaso de Osteodistrofia RenalMariana MartínezBelum ada peringkat
- Test HamiltonDokumen5 halamanTest HamiltonMariana MartínezBelum ada peringkat
- Hábitos Que Dan Forma Al Carácter CristianoDokumen4 halamanHábitos Que Dan Forma Al Carácter CristianoGuillermo PeñaBelum ada peringkat
- Lorca MesinaDokumen147 halamanLorca Mesinamgarcia_244635Belum ada peringkat
- Informe 3 - Circuitos 3Dokumen6 halamanInforme 3 - Circuitos 3Daniel J. LopezBelum ada peringkat
- Mototrailla y VolquetasDokumen39 halamanMototrailla y VolquetasDaniela Aguirre100% (1)
- Vigas de ConcretoDokumen8 halamanVigas de ConcretoJuan NimaBelum ada peringkat
- S3 Electrónica IndustrialDokumen10 halamanS3 Electrónica Industrialjunior baltazar coronado martinezBelum ada peringkat
- 201101311733090.anamnesis Llenado DigitalDokumen5 halaman201101311733090.anamnesis Llenado Digitalkarlosprofe100% (1)
- Tesis FORESTAL PDFDokumen71 halamanTesis FORESTAL PDFReynaldo LPBelum ada peringkat
- Capitulo 4.2 El ReaseguroDokumen8 halamanCapitulo 4.2 El ReaseguroRenny Isabel Silvestre De La CruzBelum ada peringkat
- Analisis de La Abeja HaraganaDokumen10 halamanAnalisis de La Abeja HaraganaSaray Beita JzBelum ada peringkat
- Gusmán, Luis - La Casa Del Dios Oculto (PDF)Dokumen100 halamanGusmán, Luis - La Casa Del Dios Oculto (PDF)John CarterBelum ada peringkat
- Syllabus Del Curso Investigación en Ciencias SocialesDokumen14 halamanSyllabus Del Curso Investigación en Ciencias SocialesMaira Alejandra Robles DuranBelum ada peringkat
- Economía 1Dokumen2 halamanEconomía 1JosephBelum ada peringkat
- Memoria Descriptiva EMAPADokumen10 halamanMemoria Descriptiva EMAPAGustavo Xavier Hidalgo Vasquez100% (1)
- Tema 4 Acepta A Jesús Como Tu SalvadorDokumen2 halamanTema 4 Acepta A Jesús Como Tu SalvadorsaulBelum ada peringkat
- Normas de Elaboración de Proyectos Del Instituto Universitario Tecnológico de Los LlanosDokumen27 halamanNormas de Elaboración de Proyectos Del Instituto Universitario Tecnológico de Los LlanosAnnys Rocio PerezBelum ada peringkat
- Depurador y SeparadorDokumen2 halamanDepurador y SeparadorJessica Alejandra TGBelum ada peringkat
- Antibioticos B-LactamicosDokumen12 halamanAntibioticos B-Lactamicosdarvin zambranoBelum ada peringkat
- Tfg-N. 1498Dokumen79 halamanTfg-N. 1498Gerard Labernia TomasBelum ada peringkat
- Weefim EspanolDokumen3 halamanWeefim EspanolPaulina Marquez RojasBelum ada peringkat
- Desarrollo Psicosocial en La Niñez MediaDokumen3 halamanDesarrollo Psicosocial en La Niñez MediaAlfredo EstradaBelum ada peringkat
- Baby Shower NuevoDokumen14 halamanBaby Shower NuevoTershe MizamoBelum ada peringkat
- Estructura Atomica PDFDokumen11 halamanEstructura Atomica PDFAinhara CalzadaBelum ada peringkat
- Tiempo de IncertidumbreDokumen164 halamanTiempo de IncertidumbretodocatsolucionesBelum ada peringkat
- MAICILLOSDokumen5 halamanMAICILLOSCarlos Rocha FernandezBelum ada peringkat
- Vision de EternidadDokumen2 halamanVision de EternidadJesus María Pastoral Argentina Uruguay Jesus MariaBelum ada peringkat