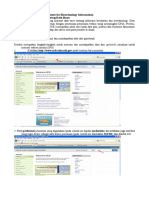
Anda mungkin juga menyukai
- Laporan Praktikum Kimia Komputasi Virtual Screening-DikonversiDokumen17 halamanLaporan Praktikum Kimia Komputasi Virtual Screening-DikonversiTiktok SerBelum ada peringkat
- Pengenalan NcbiDokumen35 halamanPengenalan Ncbitia100% (1)
- Metode Desain VaksinDokumen7 halamanMetode Desain VaksinFunikawati BaldiyahBelum ada peringkat
- GABRIELLE NitipDokumen9 halamanGABRIELLE NitipGABRIELLE AHMAD MASZATUBelum ada peringkat
- Protokol Pelatihan BioinformatikaDokumen21 halamanProtokol Pelatihan BioinformatikaI EBelum ada peringkat
- Aditra, 9-15Dokumen7 halamanAditra, 9-15RaniBelum ada peringkat
- Mengenal NCBIDokumen22 halamanMengenal NCBIrony kundaBelum ada peringkat
- 11111Dokumen8 halaman11111DyahBelum ada peringkat
- Putri UAS Bioinformatika.Dokumen2 halamanPutri UAS Bioinformatika.Salsabila HendroBelum ada peringkat
- M Bagus Firdaus - Lapsem Biomolekuler Materi 6Dokumen12 halamanM Bagus Firdaus - Lapsem Biomolekuler Materi 6Muhammad BagusBelum ada peringkat
- Galan Rizqi Yanuar - 11181017 - Laporan Praktikum Kimia Komputasi Modul VISUALISASI STRUKTUR MAKROMOLEKULDokumen12 halamanGalan Rizqi Yanuar - 11181017 - Laporan Praktikum Kimia Komputasi Modul VISUALISASI STRUKTUR MAKROMOLEKULGalan Rizqi YanuarBelum ada peringkat
- Acara 3 BioinformatikaDokumen4 halamanAcara 3 Bioinformatikaanastasia sylviankaBelum ada peringkat
- Laporan BioinformatikaDokumen17 halamanLaporan Bioinformatikamayka putraBelum ada peringkat
- Pangkalan Data Dan Mencari Serta Membaca Data Biologi Pada Gene BankDokumen9 halamanPangkalan Data Dan Mencari Serta Membaca Data Biologi Pada Gene BankGerfindo WatugigirBelum ada peringkat
- UEU Course 9828 7 - 00249Dokumen86 halamanUEU Course 9828 7 - 00249Dhea AnandaBelum ada peringkat
- Marselina Nedja - 201FF04025 - Laporan Modul 5Dokumen10 halamanMarselina Nedja - 201FF04025 - Laporan Modul 5marselina nedjaBelum ada peringkat
- WA0073. - DieditDokumen14 halamanWA0073. - Diedit006ERY MARIAMBelum ada peringkat
- Makalah Ibu Dewi Kel 6Dokumen12 halamanMakalah Ibu Dewi Kel 6Rindi Antika FamilyBelum ada peringkat
- Uas Genetika Mita Ramadhani 4213151017 Pipa21aDokumen8 halamanUas Genetika Mita Ramadhani 4213151017 Pipa21aMita RamadhaniBelum ada peringkat
- Cinta Skripsi Banget-46-54Dokumen9 halamanCinta Skripsi Banget-46-54Fyan VergarraBelum ada peringkat
- Suprianto Proposal Perbaikan 5Dokumen29 halamanSuprianto Proposal Perbaikan 5UpickBelum ada peringkat
- Petunjuk Praktikum BiofarmasetikaDokumen151 halamanPetunjuk Praktikum Biofarmasetikatania.audria.sutopoBelum ada peringkat
- 3854 7868 1 SMDokumen12 halaman3854 7868 1 SMGisela GloryBelum ada peringkat
- Bioinformatika SBG Ilmu Bantu Biologi MolekulerDokumen30 halamanBioinformatika SBG Ilmu Bantu Biologi MolekulerAswan PoranteBelum ada peringkat
- Laporan 3Dokumen9 halamanLaporan 3YasaKaryadaBelum ada peringkat
- Laporan Fix BiotekDokumen32 halamanLaporan Fix BiotekFitri Nurussani AuliaBelum ada peringkat
- LP - Biotek A2-1 Analisis BioinformatikaDokumen23 halamanLP - Biotek A2-1 Analisis BioinformatikaImtiyas AdzraBelum ada peringkat
- Axxx13 Petunjuk Praktikum Dasar Bioinformatika Genap 1819Dokumen24 halamanAxxx13 Petunjuk Praktikum Dasar Bioinformatika Genap 181931. Rizky Ade wardanaBelum ada peringkat
- Protein Engineering - Siti JulaihaDokumen189 halamanProtein Engineering - Siti JulaihaSiti Julaiha Grübner100% (1)
- Zahra Putri Aulia (B2A019004) - Analisis Desain Primer Gen COX-2 Pada Oncorhynchus MykissDokumen16 halamanZahra Putri Aulia (B2A019004) - Analisis Desain Primer Gen COX-2 Pada Oncorhynchus Mykisshyukra04Belum ada peringkat
- Acara V - IxDokumen59 halamanAcara V - IxFahrez AnsariBelum ada peringkat
- Modul NCBI Introduction BLAST 3D 10 AprilDokumen16 halamanModul NCBI Introduction BLAST 3D 10 AprilsarijuicyBelum ada peringkat
- Pengenalan Website BioinformatikaDokumen6 halamanPengenalan Website BioinformatikaDevita SariBelum ada peringkat
- Buku Petunjuk Drylab Biosel 2010Dokumen14 halamanBuku Petunjuk Drylab Biosel 2010AzhaBelum ada peringkat
- Protein Modeling Dan Pengaruh Mutasi Terhadap Struktur ProteinDokumen9 halamanProtein Modeling Dan Pengaruh Mutasi Terhadap Struktur ProteinhannayediyaBelum ada peringkat
- Bio Inform A TikaDokumen27 halamanBio Inform A TikaHeppy Setya primaBelum ada peringkat
- Bioinformatika SBG Ilmu Bantu Biologi MolekulerDokumen29 halamanBioinformatika SBG Ilmu Bantu Biologi Molekuleragnes ilyasBelum ada peringkat
- Homology ModellingDokumen28 halamanHomology Modellingwildan abiyyiBelum ada peringkat
- Tugas Baca Bioinformatika Biosel Winda Ayu Fietri (20177023)Dokumen4 halamanTugas Baca Bioinformatika Biosel Winda Ayu Fietri (20177023)windaBelum ada peringkat
- (Praktikan) Tugas Pendahuluan MODUL7Dokumen2 halaman(Praktikan) Tugas Pendahuluan MODUL7erfanus janeBelum ada peringkat
- Homology Modelling NP - 001005862 - 1p - Arg866GlyDokumen26 halamanHomology Modelling NP - 001005862 - 1p - Arg866Glywildan abiyyiBelum ada peringkat
- Proteins PDFDokumen6 halamanProteins PDFFitriBelum ada peringkat
- Review Alat Alat Lab LsihDokumen15 halamanReview Alat Alat Lab LsihRyan Nugraha100% (1)
- Praktikum Ii Pengenalan Situs BioinformaDokumen14 halamanPraktikum Ii Pengenalan Situs BioinformaAnkerz15Belum ada peringkat
- Makalah BiomolekulDokumen6 halamanMakalah BiomolekulJuliana SihombingBelum ada peringkat
- Ariefwitarto Didimensi Februari2003Dokumen6 halamanAriefwitarto Didimensi Februari2003sukmawati DamitiBelum ada peringkat
- 055 - Abdullah Khilmi - BIOTEKNOLOGI 6D - RESUME BAB 12Dokumen4 halaman055 - Abdullah Khilmi - BIOTEKNOLOGI 6D - RESUME BAB 12abdullah khilmiBelum ada peringkat
- Makalah Biologi Molekuler BioinformatikaDokumen11 halamanMakalah Biologi Molekuler BioinformatikaJosua GigirBelum ada peringkat
- LAPORAN PRAKTIKUM ANALISIS BIOMOLEKULAR Isolasi ProteinDokumen4 halamanLAPORAN PRAKTIKUM ANALISIS BIOMOLEKULAR Isolasi ProteinWahyuni WulansariBelum ada peringkat
- A0A917LGX0 - Kelompok (4) - AlphaFoldDokumen3 halamanA0A917LGX0 - Kelompok (4) - AlphaFoldDyahrosa PranataBelum ada peringkat
- Septi 2019Dokumen8 halamanSepti 2019Monyet...Belum ada peringkat
- Bab IDokumen17 halamanBab IIntan PurmalaBelum ada peringkat
- Memanfaatkan DatabaseDokumen14 halamanMemanfaatkan DatabaseAbdul Rosyid Al MuhammadyBelum ada peringkat
- Tugas Individu Mata Kuliah Analisis MakananDokumen17 halamanTugas Individu Mata Kuliah Analisis MakananTies Titis WidowatiBelum ada peringkat
- MAKALAH ImunopresipitasiDokumen11 halamanMAKALAH ImunopresipitasiAidia Latifatul Fajeria100% (2)
- M Bagus Firdaus - Lapsem Biomolekuler Materi 8Dokumen15 halamanM Bagus Firdaus - Lapsem Biomolekuler Materi 8Muhammad BagusBelum ada peringkat
- Bioteknologi 6Dokumen2 halamanBioteknologi 6alexandra misel kristieBelum ada peringkat
- Tugas 3 FITRI KURNIA - 2250397014Dokumen8 halamanTugas 3 FITRI KURNIA - 2250397014Fitri KurniaBelum ada peringkat
- Analisis - Protein - Biokimm NURULDokumen17 halamanAnalisis - Protein - Biokimm NURULNurul AiniBelum ada peringkat