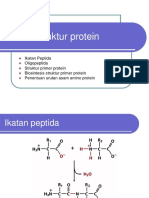
Anda mungkin juga menyukai
- Tugas 1 Biokimia PanganDokumen7 halamanTugas 1 Biokimia PanganLimaran sariBelum ada peringkat
- Astrid Alfira Noermawati - 20030234024 - Tugas Kimfis LKM MOTDokumen19 halamanAstrid Alfira Noermawati - 20030234024 - Tugas Kimfis LKM MOTAstrid AlfiraBelum ada peringkat
- Asam Amino-ProteinDokumen41 halamanAsam Amino-ProteinAnonymous YbWxYhphrRBelum ada peringkat
- Asam Amino Dan ProteinDokumen51 halamanAsam Amino Dan ProteinHendy PutraBelum ada peringkat
- Struktur 3 D ProteinDokumen10 halamanStruktur 3 D ProteinDita FitrianiBelum ada peringkat
- Struktur & Fungsi Protein-2016Dokumen71 halamanStruktur & Fungsi Protein-2016Mutia KeumalahayatiBelum ada peringkat
- Bagian VI Struktur Tiga Dimensi ProteinDokumen35 halamanBagian VI Struktur Tiga Dimensi ProteinmarfuzahBelum ada peringkat
- Tugas 1 PANG4222Dokumen3 halamanTugas 1 PANG4222AgungBelum ada peringkat
- Struktur Molekul ProteinDokumen11 halamanStruktur Molekul ProteinNia Sasria IdrisBelum ada peringkat
- Kimia Organik ProteinDokumen33 halamanKimia Organik ProteinAdi Suryadi PutraBelum ada peringkat
- Struktur Molekul ProteinDokumen10 halamanStruktur Molekul ProteinSiska Winti SoneBelum ada peringkat
- Makromolekul BiologiDokumen4 halamanMakromolekul BiologiRatna PermatasariBelum ada peringkat
- Rangkuman Lab Biomedic 2Dokumen10 halamanRangkuman Lab Biomedic 2TriyaBelum ada peringkat
- QBD PMIB Cheri, Rizka, JulianDokumen44 halamanQBD PMIB Cheri, Rizka, JulianDeasyka YBelum ada peringkat
- 02 ProteinDokumen32 halaman02 ProteinSyahilaBelum ada peringkat
- BiokimfisDokumen16 halamanBiokimfisRuth Michelia SavitriBelum ada peringkat
- Struktur Fungsi ProteinDokumen47 halamanStruktur Fungsi ProteinNatsu DragneelBelum ada peringkat
- Protein Serat Digolongkan Menjadi 2 Jenis UtamaDokumen4 halamanProtein Serat Digolongkan Menjadi 2 Jenis UtamaR.S.G.FBelum ada peringkat
- Prinsip Dasar Desain Obat II (Struktur Dan Sifat Molekul Target)Dokumen47 halamanPrinsip Dasar Desain Obat II (Struktur Dan Sifat Molekul Target)MISSYE DAYANA SABILLABelum ada peringkat
- Bab 5. Protein - Struktur Ordo TinggiDokumen12 halamanBab 5. Protein - Struktur Ordo TinggiTeuku Ilham AkbarBelum ada peringkat
- Asam AminoDokumen31 halamanAsam AminoFiryal Fairuztsana NugrahaBelum ada peringkat
- Bio ProteinDokumen11 halamanBio ProteinREBECCA REGINA ELVINA BUDIONOBelum ada peringkat
- Lkps Materi Genetik 2Dokumen14 halamanLkps Materi Genetik 2Tim 1FRBelum ada peringkat
- LTM Biomol 2Dokumen10 halamanLTM Biomol 2Hasbi AsshidiqiBelum ada peringkat
- Materi Genetik 2Dokumen15 halamanMateri Genetik 2Tim 1FRBelum ada peringkat
- EnzimDokumen8 halamanEnzimEka PujiastutiBelum ada peringkat
- Struktur Organel SelDokumen12 halamanStruktur Organel SelRIFAM zBelum ada peringkat
- 6.materi, Soal Gen Kanggo SliramuDokumen37 halaman6.materi, Soal Gen Kanggo SliramuNabila AnjaniBelum ada peringkat
- ProteinDokumen14 halamanProteinNaufan Nurrosyid PBelum ada peringkat
- Fungsi ProteinDokumen48 halamanFungsi ProteinYuliana BaanBelum ada peringkat
- BIOKIM TUMBUHAN 8 (PROT & ENZIM) Pak SaryonoDokumen91 halamanBIOKIM TUMBUHAN 8 (PROT & ENZIM) Pak SaryonoLulu Angela NirahaiBelum ada peringkat
- Struktur ProteinDokumen53 halamanStruktur ProteinmaudiBelum ada peringkat
- Identifikasi Asam Amino Dan ProteinDokumen38 halamanIdentifikasi Asam Amino Dan ProteinAinuzzahraBelum ada peringkat
- Biologi Molekuler Percobaan 3Dokumen7 halamanBiologi Molekuler Percobaan 3Neni Yuli SulistianiBelum ada peringkat
- 3 - Asam Amino & ProteinDokumen54 halaman3 - Asam Amino & ProteinLisna WadampakaBelum ada peringkat
- Asam Amino Dan Protein NewDokumen60 halamanAsam Amino Dan Protein Newyoustiana dwi rusitaBelum ada peringkat
- Metabolisme Protein Dan Asam AminoDokumen31 halamanMetabolisme Protein Dan Asam AminoSaphiraEvaniBelum ada peringkat
- Asam Amino, Peptida Dan ProteinDokumen33 halamanAsam Amino, Peptida Dan ProteinRiska Afrillia Cipti AndiniBelum ada peringkat
- Diskusi 2 Biokimia PanganDokumen3 halamanDiskusi 2 Biokimia PanganTania AprilianaBelum ada peringkat
- Rivaldi Pratama - H041201034 - Tugas 1 BiokimiaDokumen8 halamanRivaldi Pratama - H041201034 - Tugas 1 BiokimiaRivaldi PratamaBelum ada peringkat
- LKPD 3.3 Pert 2Dokumen6 halamanLKPD 3.3 Pert 2Rafvena FilonaPutriBelum ada peringkat
- Pertemuan 6. Asam NukleatDokumen63 halamanPertemuan 6. Asam NukleatFatmaBelum ada peringkat
- Polipeptida Dan ProteinDokumen9 halamanPolipeptida Dan ProteinFikriansyahBelum ada peringkat
- Struktur Asam Nukleat - PangiastikaDokumen34 halamanStruktur Asam Nukleat - PangiastikapangiastikaBelum ada peringkat
- Struktur Dan Fungsi Protein - ChibiDokumen28 halamanStruktur Dan Fungsi Protein - ChibiAnanda SelaBelum ada peringkat
- Aliran Informasi GenetikDokumen13 halamanAliran Informasi GenetikIts'Elzz SBelum ada peringkat
- LKPD Materi Genetik 2Dokumen13 halamanLKPD Materi Genetik 2Tim 1FRBelum ada peringkat
- Konsep GenetikaDokumen9 halamanKonsep GenetikaFaisna FirnandoBelum ada peringkat
- Asam AminoDokumen11 halamanAsam AminoGokil Heru80% (5)
- Pengantar BiokimiaDokumen23 halamanPengantar BiokimiaDesi Triutami SalehBelum ada peringkat
- Perbedaan Enzim Dan ProteinDokumen2 halamanPerbedaan Enzim Dan Proteinanis martiaBelum ada peringkat
- Cell Structure FunctionDokumen61 halamanCell Structure FunctionKaniaBelum ada peringkat
- Protoplasma PDFDokumen52 halamanProtoplasma PDFDaniel Victor100% (1)
- Materi 4 Protein (1) RevDokumen24 halamanMateri 4 Protein (1) RevMawar stevani sitanggangBelum ada peringkat
- Asam NukleatDokumen5 halamanAsam Nukleatzhafran kamalBelum ada peringkat
- Kelainan Sturuktur Dan Fungsi RibosomDokumen30 halamanKelainan Sturuktur Dan Fungsi RibosomBrowniepie50% (2)
- Tugas Pertemuan 5 SFBDokumen10 halamanTugas Pertemuan 5 SFBSafitri NurulBelum ada peringkat
- Perbedaan Enzim Dan ProteinDokumen2 halamanPerbedaan Enzim Dan ProteinAhmad Jamaludin100% (1)
- ProteinDokumen134 halamanProteinnisa100% (1)
- Evaluasi Tablet Salut FilmDokumen15 halamanEvaluasi Tablet Salut FilmGalan Rizqi YanuarBelum ada peringkat
- PerhitunganDokumen17 halamanPerhitunganGalan Rizqi YanuarBelum ada peringkat
- Galan Rizqi Yanuar - 11181017 - Laporan Metode Statistik Pertemuan 10 Uji AnovaDokumen4 halamanGalan Rizqi Yanuar - 11181017 - Laporan Metode Statistik Pertemuan 10 Uji AnovaGalan Rizqi YanuarBelum ada peringkat
- Kuis Pertemuan 9Dokumen2 halamanKuis Pertemuan 9Galan Rizqi YanuarBelum ada peringkat
- Uji Stabilitas (: Praktikum Farmasi Fisika 2)Dokumen7 halamanUji Stabilitas (: Praktikum Farmasi Fisika 2)Galan Rizqi YanuarBelum ada peringkat
- Deny Puriyani Azhary, M.Si., AptDokumen10 halamanDeny Puriyani Azhary, M.Si., AptGalan Rizqi YanuarBelum ada peringkat
- Galan Rizqi Yanuar - 11181017 - Laporan Praktikum Kimia Komputasi Modul VISUALISASI STRUKTUR MAKROMOLEKULDokumen12 halamanGalan Rizqi Yanuar - 11181017 - Laporan Praktikum Kimia Komputasi Modul VISUALISASI STRUKTUR MAKROMOLEKULGalan Rizqi YanuarBelum ada peringkat