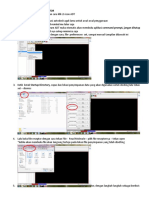
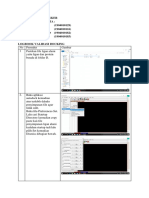
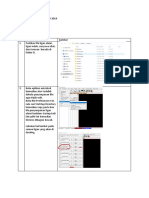
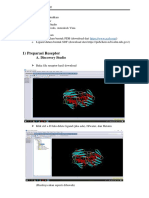
Anda mungkin juga menyukai
- Tutorial AutodockDokumen11 halamanTutorial AutodockA.S.H projecBelum ada peringkat
- Logbook Kimed IiDokumen11 halamanLogbook Kimed IiTatang SupriatnaBelum ada peringkat
- LOGBOOK KELOMPOK ANTIKANKER VALIDASI DOCKING-dikonversiDokumen12 halamanLOGBOOK KELOMPOK ANTIKANKER VALIDASI DOCKING-dikonversiNamira NaaziahBelum ada peringkat
- Tutorial Docking AutodockDokumen3 halamanTutorial Docking AutodockFuzi Fauzia Fajrin Fauzia FajrinBelum ada peringkat
- Logbook Docking Senyawa Ligan Alami, Ligan Induk, Ligan Turunan Dan Senyawa Obat - Kelampok 6 Farmasi D 2019Dokumen10 halamanLogbook Docking Senyawa Ligan Alami, Ligan Induk, Ligan Turunan Dan Senyawa Obat - Kelampok 6 Farmasi D 2019Elis Nurul IkhlasBelum ada peringkat
- Tutorial Docking Autodock4.2 (New)Dokumen3 halamanTutorial Docking Autodock4.2 (New)MochamadFathurrBelum ada peringkat
- Cara Docking Senyawa Dengan Autodock 4Dokumen10 halamanCara Docking Senyawa Dengan Autodock 4Carlos RumangunBelum ada peringkat
- Panduan Molecular Docking Autodock 4.0Dokumen17 halamanPanduan Molecular Docking Autodock 4.0alisia difianaBelum ada peringkat
- DOCKING FEBYANTI SAFITRI Dan GUSTI MAHARINIDokumen7 halamanDOCKING FEBYANTI SAFITRI Dan GUSTI MAHARININasrullah HamdaniBelum ada peringkat
- Modul Bioinfor DockingDokumen17 halamanModul Bioinfor DockinggitaBelum ada peringkat
- Modul Biokimia Proyek 2: Molecular DockingDokumen31 halamanModul Biokimia Proyek 2: Molecular DockingNISRINA NAJLA HUWAIDABelum ada peringkat
- LaporanDokumen13 halamanLaporanLia NurhayatiBelum ada peringkat
- Mari Belajar Molecular DockingDokumen5 halamanMari Belajar Molecular DockingAnggie WindrianiBelum ada peringkat
- Tutorial Autodock VinaDokumen22 halamanTutorial Autodock VinapmuftiaBelum ada peringkat
- Laporan Praktikum Molecular DockingDokumen14 halamanLaporan Praktikum Molecular DockingIsnainiBelum ada peringkat
- Job 4 Sistem Operasi - Konfigurasi LinuxDokumen4 halamanJob 4 Sistem Operasi - Konfigurasi LinuxfadillaBelum ada peringkat
- Modul 3 - Praktikum Sistem OperasiDokumen5 halamanModul 3 - Praktikum Sistem Operasifajar mohamadBelum ada peringkat
- Laporan Praktikum Kimia KomputasiDokumen19 halamanLaporan Praktikum Kimia KomputasiSHELFI RENITABelum ada peringkat
- Modul Ketiga Praktikum Sistem OperasiDokumen8 halamanModul Ketiga Praktikum Sistem Operasilegi afatahBelum ada peringkat
- Turorial Molecular DockingDokumen34 halamanTurorial Molecular DockinggedeBelum ada peringkat
- Bioinformatika StepsDokumen8 halamanBioinformatika StepsAriane BeninaBelum ada peringkat
- Validasi ReseptorDokumen3 halamanValidasi Reseptorsyifa nurul ainiBelum ada peringkat
- Tutorial Molecular Docking Dengan Autodock VinaDokumen7 halamanTutorial Molecular Docking Dengan Autodock Vinaludya pulungBelum ada peringkat
- Additional ModulDokumen14 halamanAdditional ModulYudi PratamaBelum ada peringkat
- Tutorial Autodock 4.2 - 2018Dokumen6 halamanTutorial Autodock 4.2 - 2018lokawati muliaBelum ada peringkat
- Tutorial Docking Andi BasoDokumen7 halamanTutorial Docking Andi BasoHIDAYATBelum ada peringkat
- Modul Sendiri LolDokumen33 halamanModul Sendiri LolyaminBelum ada peringkat
- KELOMPOK ANTIBAKTERI-ligan Dan Reseptor-Farmasi 3dDokumen9 halamanKELOMPOK ANTIBAKTERI-ligan Dan Reseptor-Farmasi 3delicenrlBelum ada peringkat
- Tutorial Simulasi Molecular Docking Menggunakan DOCK6Dokumen6 halamanTutorial Simulasi Molecular Docking Menggunakan DOCK6putryaBelum ada peringkat
- Laporan Konfigurasi Linux Sistem OperasiDokumen5 halamanLaporan Konfigurasi Linux Sistem OperasiNazla AnastashaBelum ada peringkat
- Alur DockingDokumen1 halamanAlur DockingAnnisa Zesika DewiBelum ada peringkat
- Kimia MedisinalDokumen19 halamanKimia MedisinalAnjali WidhiyaniBelum ada peringkat
- Laporan Praktikum Judul Konfigurasi Linu 3Dokumen12 halamanLaporan Praktikum Judul Konfigurasi Linu 3legi afatahBelum ada peringkat
- Modul IVDokumen3 halamanModul IVHikmatun NazilaBelum ada peringkat
- Yunia Sara A.K 2443018237 Tugas 8 Prak - FarkompiDokumen4 halamanYunia Sara A.K 2443018237 Tugas 8 Prak - FarkompiFOLANBelum ada peringkat
- Terjemahan BAB Asli 14 12Dokumen17 halamanTerjemahan BAB Asli 14 12Rich ZeusBelum ada peringkat
- Tutorial Molecular DynamicsDokumen5 halamanTutorial Molecular DynamicsFuzi Fauzia Fajrin Fauzia FajrinBelum ada peringkat
- Sisop - Andika Rifqi Maulana - Bab7Dokumen17 halamanSisop - Andika Rifqi Maulana - Bab7Andika Rifqi MaulanaBelum ada peringkat
- Membuka Dan Menutup CDDokumen23 halamanMembuka Dan Menutup CDAlam InovatifBelum ada peringkat
- Laporan Praktikum 4Dokumen16 halamanLaporan Praktikum 4Anugrah MuktiBelum ada peringkat
- Modul #2 - Rahmanita Widiyanti - H1051171007Dokumen18 halamanModul #2 - Rahmanita Widiyanti - H1051171007rahmanita widiyantiBelum ada peringkat
- Sisop 1301218680Dokumen22 halamanSisop 1301218680alipiqriBelum ada peringkat
- Jobsheet Instalasi Proftpd Server-1Dokumen7 halamanJobsheet Instalasi Proftpd Server-1EvitaaBelum ada peringkat
- Carakerja TyyDokumen2 halamanCarakerja TyyEfanBelum ada peringkat
- Bab 3Dokumen14 halamanBab 3Rezvigel AmandaBelum ada peringkat
- HadoopDokumen3 halamanHadoopJamiatBelum ada peringkat
- Rangkuman Sistem Operasi UasDokumen22 halamanRangkuman Sistem Operasi UasGUJES JAYABelum ada peringkat
- Job 5 Sistem Operasi Kak DenyDokumen9 halamanJob 5 Sistem Operasi Kak DenydindBelum ada peringkat
- Bab9 185150301111027 HaryantosihombingDokumen23 halamanBab9 185150301111027 HaryantosihombingHaryanto SihombingBelum ada peringkat
- Laporan Praktikum DockingDokumen14 halamanLaporan Praktikum DockingMuhammadRezaPahleviBelum ada peringkat
- Kimed 6Dokumen7 halamanKimed 6rizaldi rahmatullahBelum ada peringkat
- Pertemuan 16Dokumen9 halamanPertemuan 16Angki NusiBelum ada peringkat
- Laporan Framework CakephpDokumen11 halamanLaporan Framework CakephpMuhammad RamdeniBelum ada peringkat
- MODUL PRAKTIKUM - Instalasi Tool: Tim Dosen - Algoritma & Pemrograman - Universitas Amikom YogyakartaDokumen7 halamanMODUL PRAKTIKUM - Instalasi Tool: Tim Dosen - Algoritma & Pemrograman - Universitas Amikom YogyakartaNur RizkiBelum ada peringkat
- Molecular DockingDokumen6 halamanMolecular DockingYasaKaryadaBelum ada peringkat
- Praktikum Pbo Pertemuan 7Dokumen13 halamanPraktikum Pbo Pertemuan 7tzyBelum ada peringkat
- BKPM DataWarehouseDokumen18 halamanBKPM DataWarehouseBayu SBelum ada peringkat
- Membuat Aplikasi Bisnis Menggunakan Visual Studio Lightswitch 2013Dari EverandMembuat Aplikasi Bisnis Menggunakan Visual Studio Lightswitch 2013Penilaian: 3.5 dari 5 bintang3.5/5 (7)
- Mari Belajar Pemrograman Berorientasi Objek menggunakan Visual C# 6.0Dari EverandMari Belajar Pemrograman Berorientasi Objek menggunakan Visual C# 6.0Penilaian: 4 dari 5 bintang4/5 (16)
- Io Resin Empedu FixDokumen1 halamanIo Resin Empedu FixFina RepitiyanaBelum ada peringkat
- Interaksi Obat Golongan Ezetimibe Dan NiacinDokumen2 halamanInteraksi Obat Golongan Ezetimibe Dan NiacinFina RepitiyanaBelum ada peringkat
- Interaksi Obat Golongan FibratDokumen2 halamanInteraksi Obat Golongan FibratFina RepitiyanaBelum ada peringkat
- Biofarmasetik DAN Farmakokinetik Klinik: Prof. Dr. Apt - Jessie Sofia PamudjiDokumen47 halamanBiofarmasetik DAN Farmakokinetik Klinik: Prof. Dr. Apt - Jessie Sofia PamudjiFina RepitiyanaBelum ada peringkat
- PENDAHULUAN KULIAH FARMASI INDUSTRI Apoteker NEWDokumen46 halamanPENDAHULUAN KULIAH FARMASI INDUSTRI Apoteker NEWFina RepitiyanaBelum ada peringkat
- Interaksi Obat Golongan StatinDokumen2 halamanInteraksi Obat Golongan StatinFina RepitiyanaBelum ada peringkat
- Pengaruh Penambahan Bubur Bayi Afkir Kukus Dalam Ransum Terhadap Performan Domba Lokal Jantan AbstrakDokumen45 halamanPengaruh Penambahan Bubur Bayi Afkir Kukus Dalam Ransum Terhadap Performan Domba Lokal Jantan AbstrakFina RepitiyanaBelum ada peringkat
- Format Surat Pernyataan Bebas PlagiasiDokumen1 halamanFormat Surat Pernyataan Bebas PlagiasiFina RepitiyanaBelum ada peringkat
- UntitledDokumen9 halamanUntitledFina RepitiyanaBelum ada peringkat
- Rancangan Formulasi Dan Evaluasi Bentuk Sediaan Farmasi: Materi KuliahDokumen4 halamanRancangan Formulasi Dan Evaluasi Bentuk Sediaan Farmasi: Materi KuliahFina RepitiyanaBelum ada peringkat
- Evaluasi Sediaan Hand and Body Lotion Ekstrak Etanol Daun KatukDokumen77 halamanEvaluasi Sediaan Hand and Body Lotion Ekstrak Etanol Daun KatukFina RepitiyanaBelum ada peringkat
- T1kosmetika Finarepitiyana 1804010134 F4DDokumen3 halamanT1kosmetika Finarepitiyana 1804010134 F4DFina RepitiyanaBelum ada peringkat
- Kel5 Kromatografiafinitas Farmasi4dDokumen17 halamanKel5 Kromatografiafinitas Farmasi4dFina RepitiyanaBelum ada peringkat
- Laporanakhir KKN Tematik Ilham Hilmi ADokumen24 halamanLaporanakhir KKN Tematik Ilham Hilmi AFina RepitiyanaBelum ada peringkat
- Kel 3 Simplisia Daun Jambu Farm 4DDokumen11 halamanKel 3 Simplisia Daun Jambu Farm 4DFina RepitiyanaBelum ada peringkat
- Reviewjurnal Regigriyasucipta 1804010120 F4DDokumen3 halamanReviewjurnal Regigriyasucipta 1804010120 F4DFina RepitiyanaBelum ada peringkat
- Sterilisasi - Kelompok 7 - F4DDokumen8 halamanSterilisasi - Kelompok 7 - F4DFina RepitiyanaBelum ada peringkat
- PERTEMUAN 2 Terminologi MorfologiDokumen31 halamanPERTEMUAN 2 Terminologi MorfologiFina RepitiyanaBelum ada peringkat
- LAPORAN AKHIR-FINA REPITIYANA-1804010134-KELOMPOK 40-DikonversiDokumen31 halamanLAPORAN AKHIR-FINA REPITIYANA-1804010134-KELOMPOK 40-DikonversiFina RepitiyanaBelum ada peringkat
- LAPORAN AKHIR REGI GRIYA SUCIPTA-1804010120-KELOMPOK 36-DikonversiDokumen54 halamanLAPORAN AKHIR REGI GRIYA SUCIPTA-1804010120-KELOMPOK 36-DikonversiFina RepitiyanaBelum ada peringkat
- Pertemuan 5 ThallophytaDokumen38 halamanPertemuan 5 ThallophytaFina RepitiyanaBelum ada peringkat
- DiskoneksiDokumen27 halamanDiskoneksiFina RepitiyanaBelum ada peringkat
- Pertemuan 3 Organ Tumbuhan Tingkat TinggiDokumen42 halamanPertemuan 3 Organ Tumbuhan Tingkat TinggiFina RepitiyanaBelum ada peringkat
- Pemutusan 2 Garis C-X Senyawa 1,3 DisfungsionalDokumen8 halamanPemutusan 2 Garis C-X Senyawa 1,3 DisfungsionalFina RepitiyanaBelum ada peringkat
- Reaksi OrganikDokumen77 halamanReaksi OrganikFina RepitiyanaBelum ada peringkat
- Pemutusan 2 Gugus C-X Dalam Senyawa Organik (11dan 12)Dokumen7 halamanPemutusan 2 Gugus C-X Dalam Senyawa Organik (11dan 12)Fina RepitiyanaBelum ada peringkat